CADD
原理:计算Ligand与Target蛋白的结合自由能,自由能越负值、二者结合越强。
AIDD: 从 ChEMBL 等数据库中获取化合物的生物功能(e.g.CBSIs),用以训练模型;用模型筛选更大的数据库,得到潜在的功能分子,之后进行CADD或者wet实验。
结构文件准备
Protein
RCSB PDB:输入target(e.g. HMG-CoA Reductase)或药品名称(e.g. ibuprofen, imatinib)。网页上提供3D View。
UniPortKB: 蛋白质序列数据库,也可以由PDB跳转(Go to UniPortKB);一个序列可以有多个结构(.pdb),选优即可。
- 人源性,哺乳动物亦可
- 最好有共结晶化合物(Ligands)
- Resolution尽量小(分辨率高, <=2 A, A=1e-10)
- 有权威参考文献(Source:JMC,EJMC)
- 根据需求选择不同覆盖程度的结构文件(参考 UniPortKB 中 ‘Positions’ 中覆盖,e.g. 229-515)
- 预处理:修补缺失氨基酸,添加H原子,修正氨基酸构想,考察酸碱性氨基酸的质子化状态,etc
- 若无合适结构文件,则需要预测蛋白质结构:同源/de-novo
Ligand
主要是Small Chemical,但也可以是protein
- PDB自带
- ZINC,PubChem,BindingDB,Cambridge Structural Database下载
- 在线绘制1:http://www.swissadme.ch/
- 在线绘制2: https://chemaxon.com/marvin
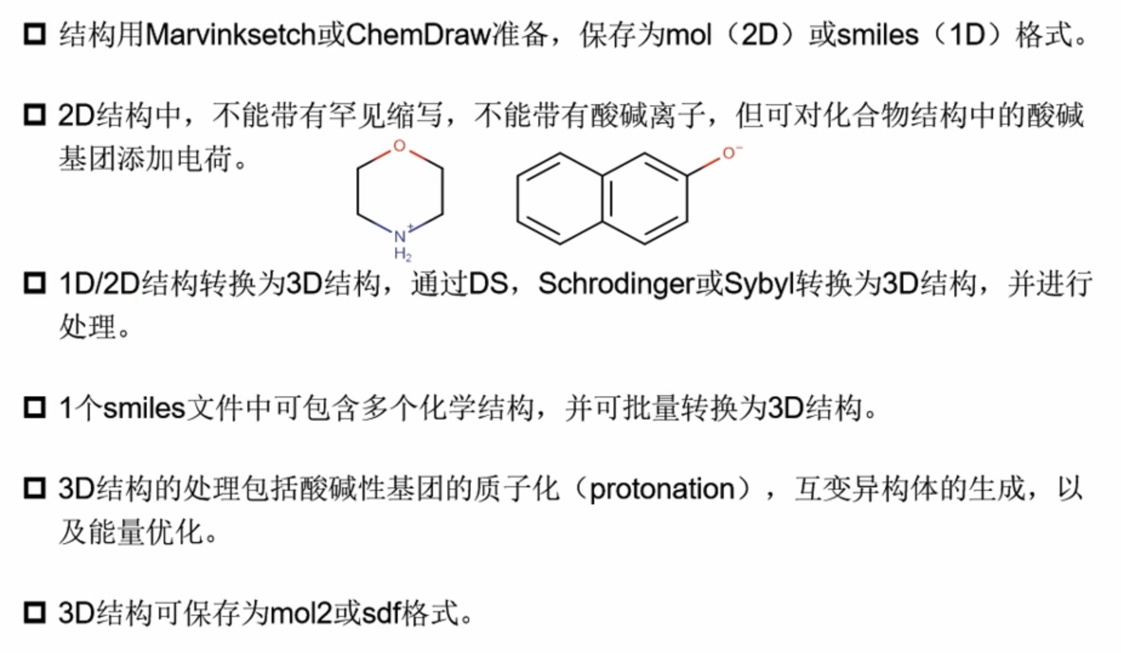
- MarvinSketch 绘制
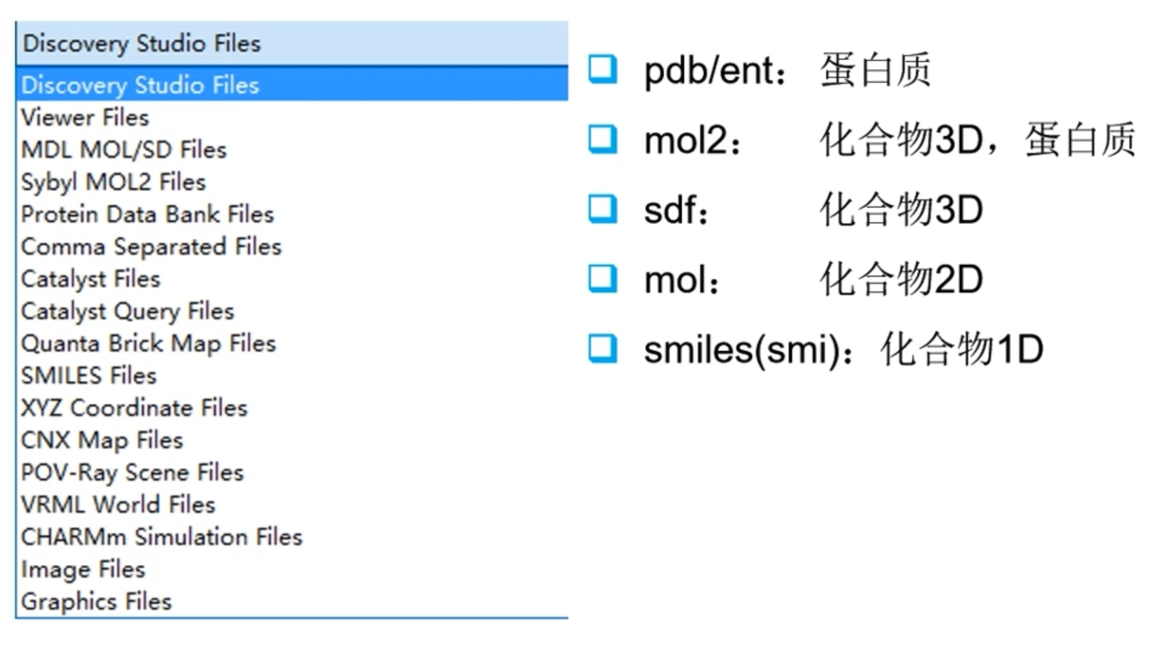
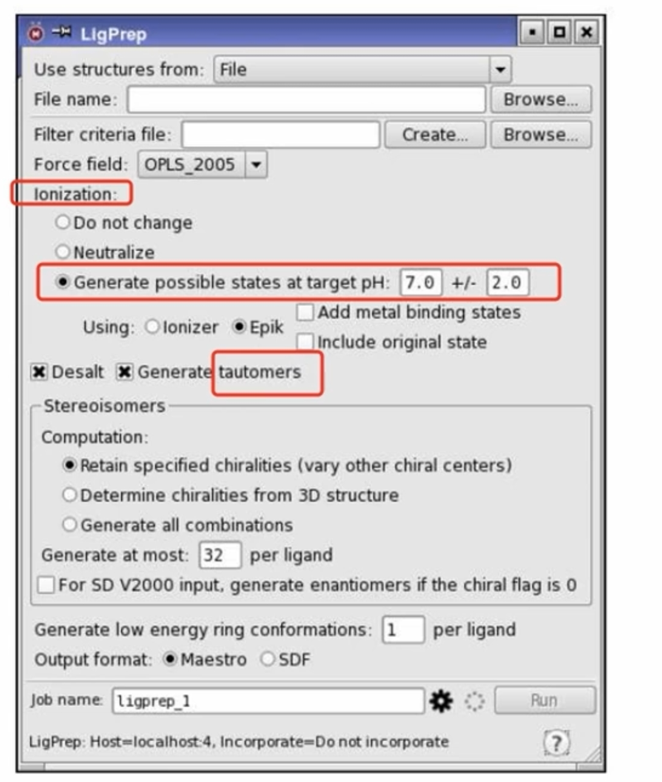
- 1D/2D可在线绘制,1D/2D to 3D的转换可使用RDKit(加H、力场优化、etc)或者Discovery Studio(Chemistry Tab 处理)、Schrödinger(LigPrep:计算化合物在活性位点的不同构象时的结合自由能)等。
可视化工具
- Discovery Studio
- PyMOL
- Maestro (Schrödinger)
- ICM Browser
- UCSF Chimera
发现先导化合物
虚拟筛选(Virtual Screening):对化合物库中大量分子进行筛选,发现结合方式、结合能量最好的先导化合物,也许具有活性。(缩小候选化合物范围)
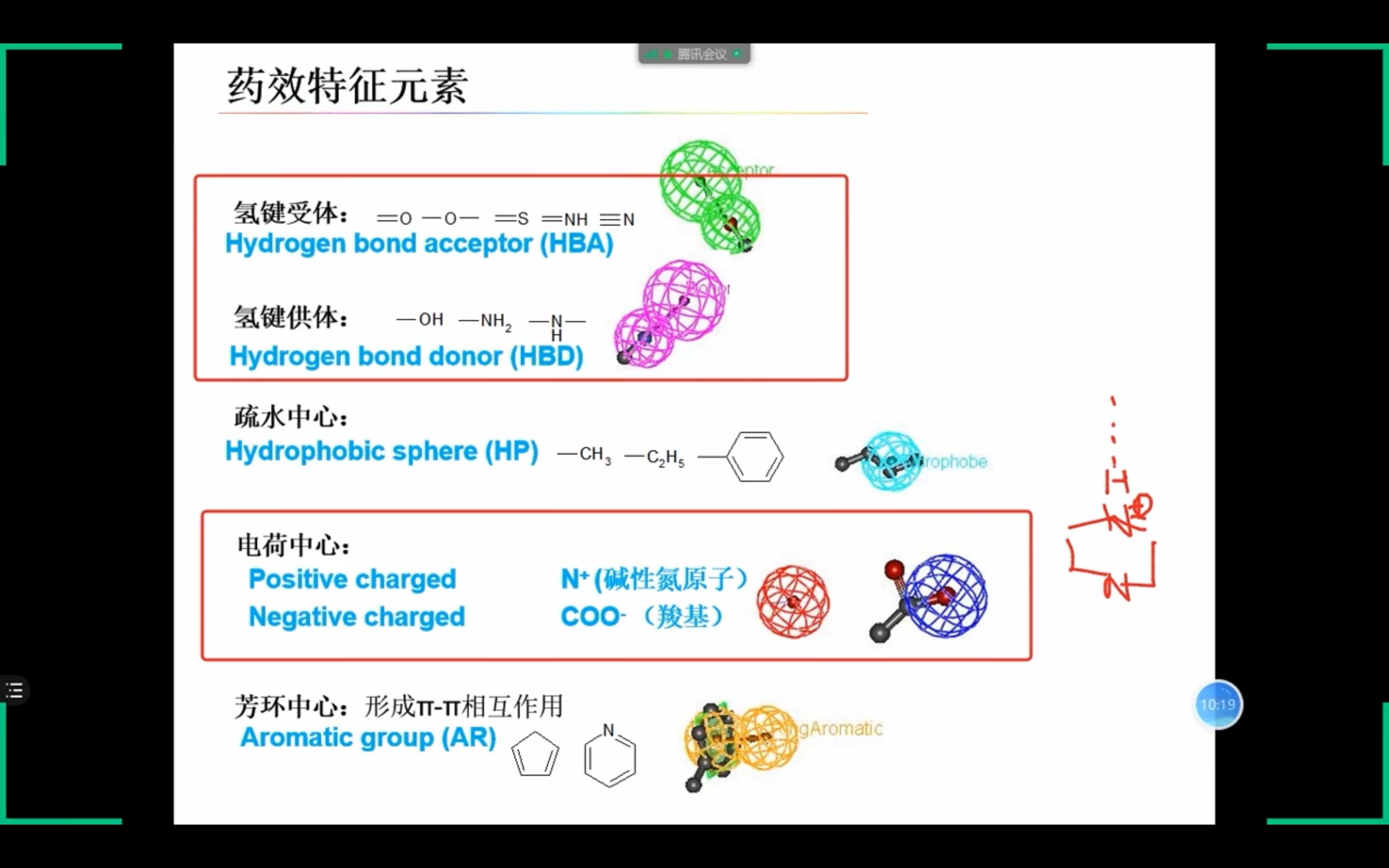
基于药效团特征
药效团(药效特征元素): 活性化合物共有的、对化合物的活性有重要影响的一组原子或基团的空间排列组合。 以下2种方法结果应一致:
- by ligand(某蛋白不同ligand的共同特征)
- by protein receptor(观察protein与ligand的binding cavity)
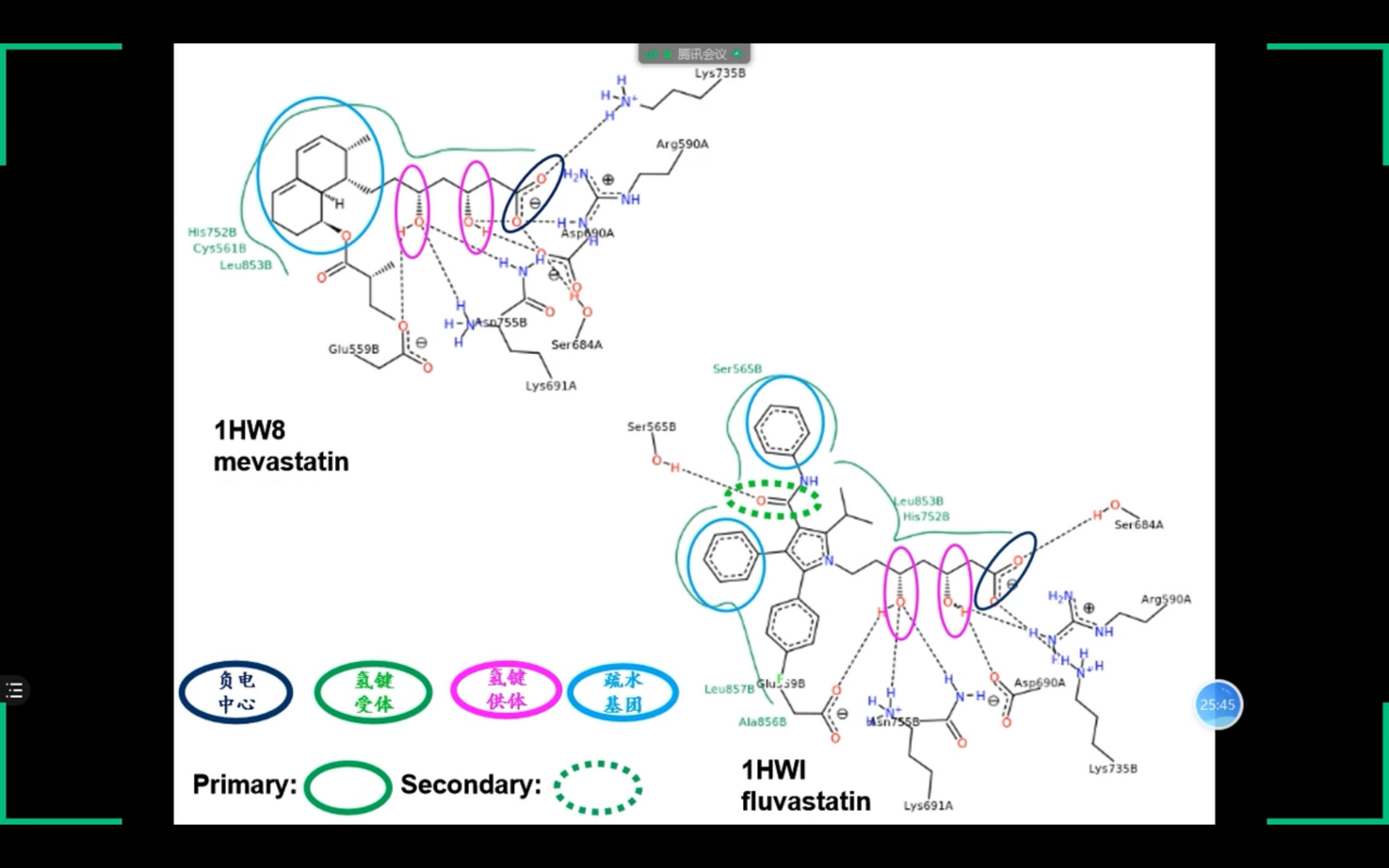
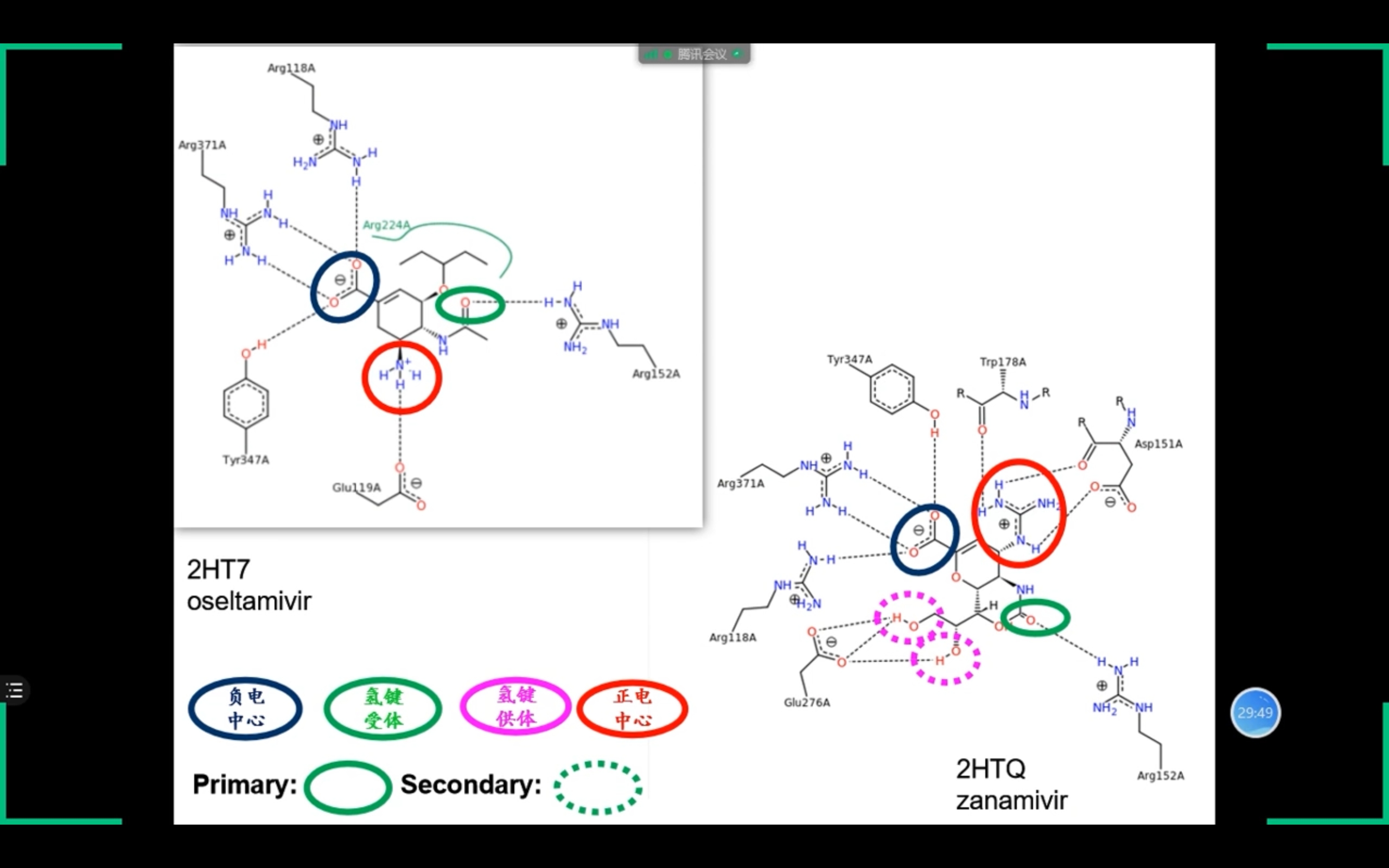
药效团发现
- Poseview(2D online): https://proteins.plus/
- PLIP(3D online): https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index
- LigPlot
- Maestro (Schrödinger)
- Discovery Studio,LigandScout(见下文筛选)
Poseview 示例
Step 1: https://proteins.plus/4dkl
Step 2: create pocket -->
Step 3: PoseView --- Settings --- Click Ligands[+] on the left to fillin 'Ligands'
Step 4: Submit Calculation
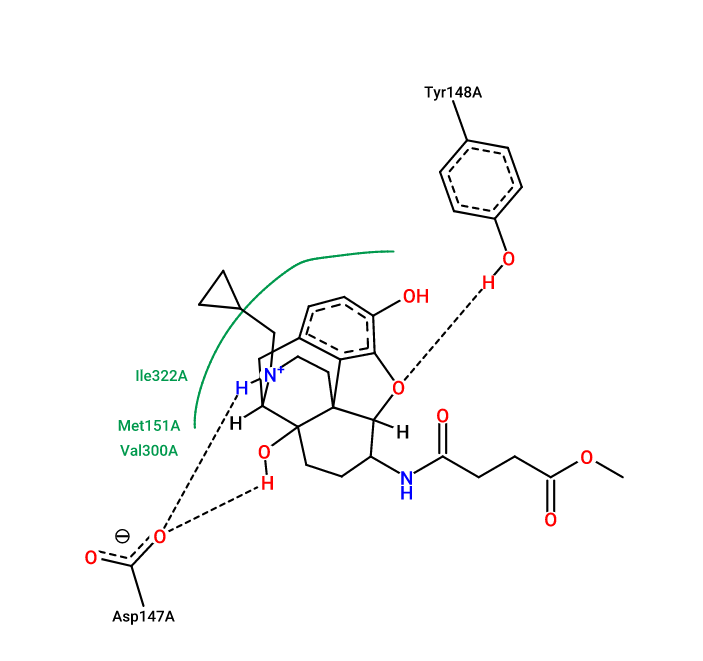
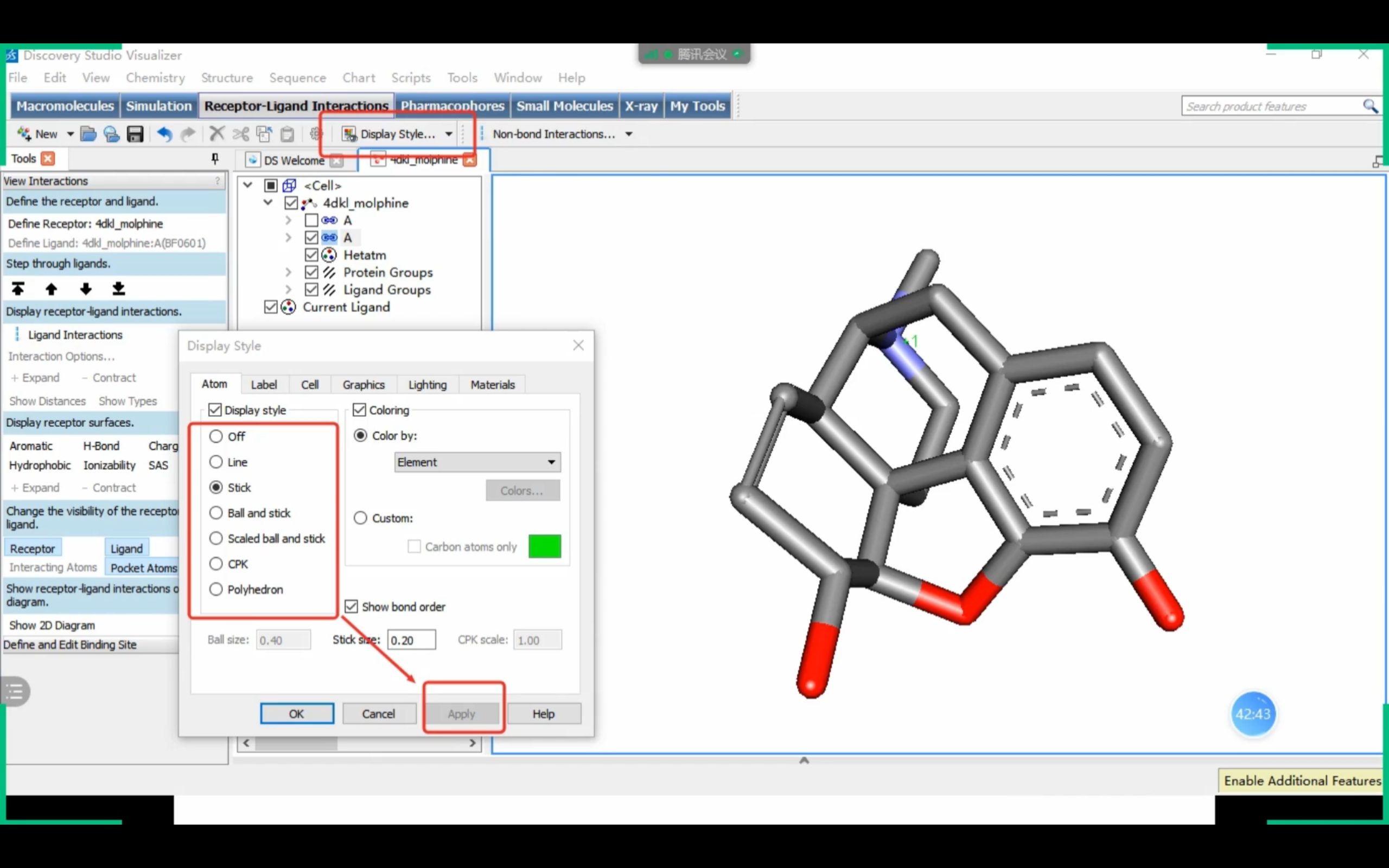
Discovery Studio
课程截图 (Receptor-Ligand Interactions)
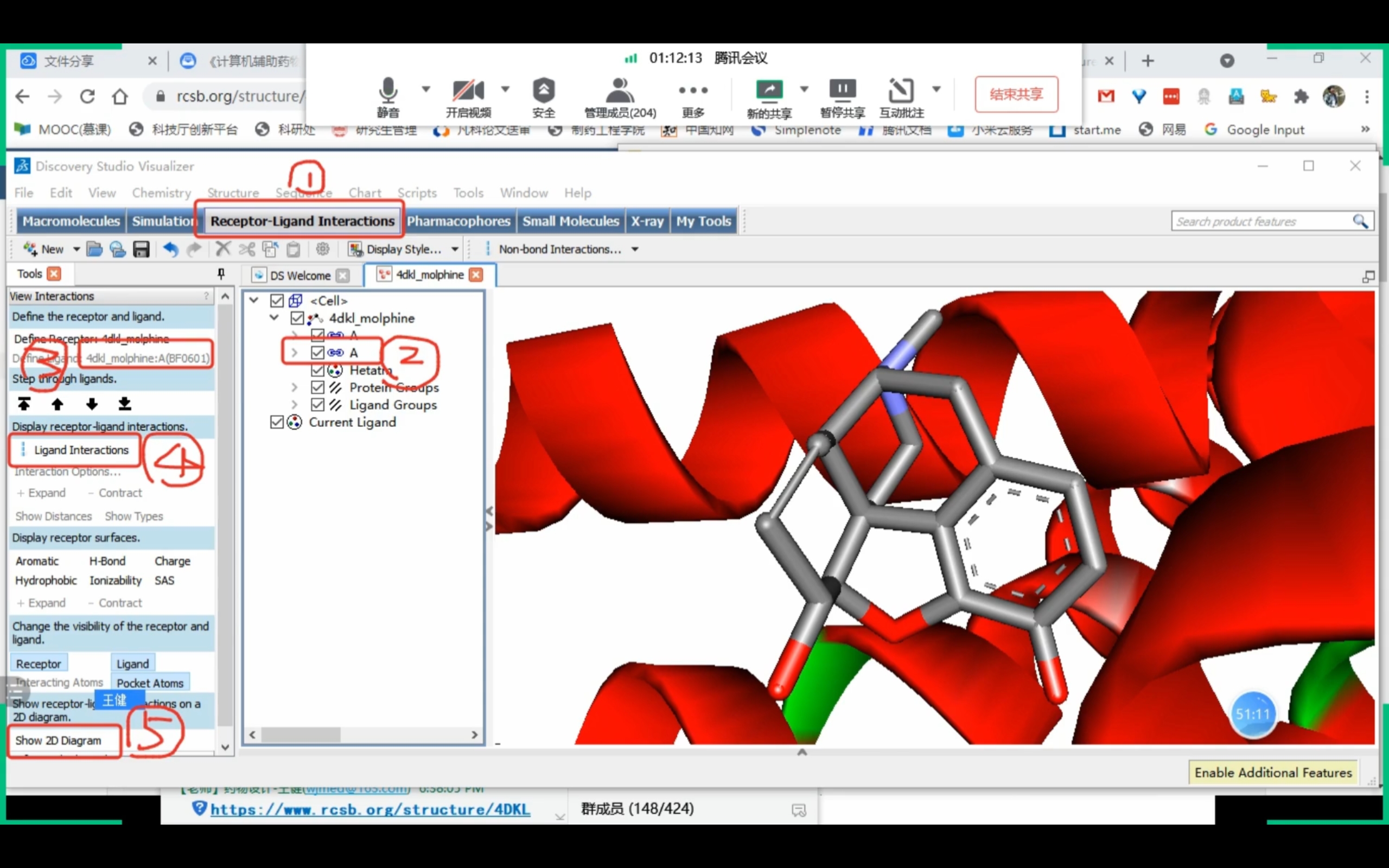
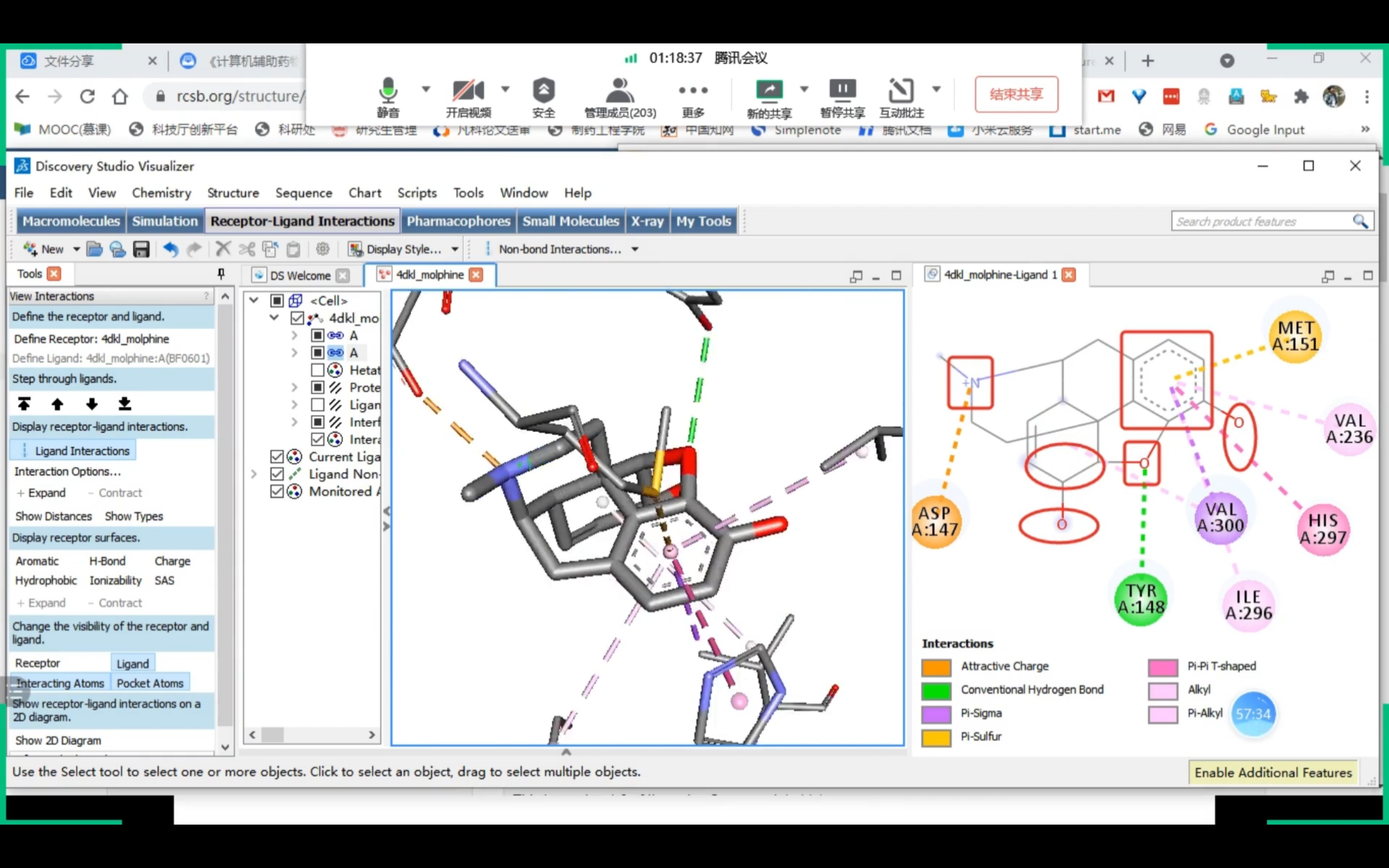
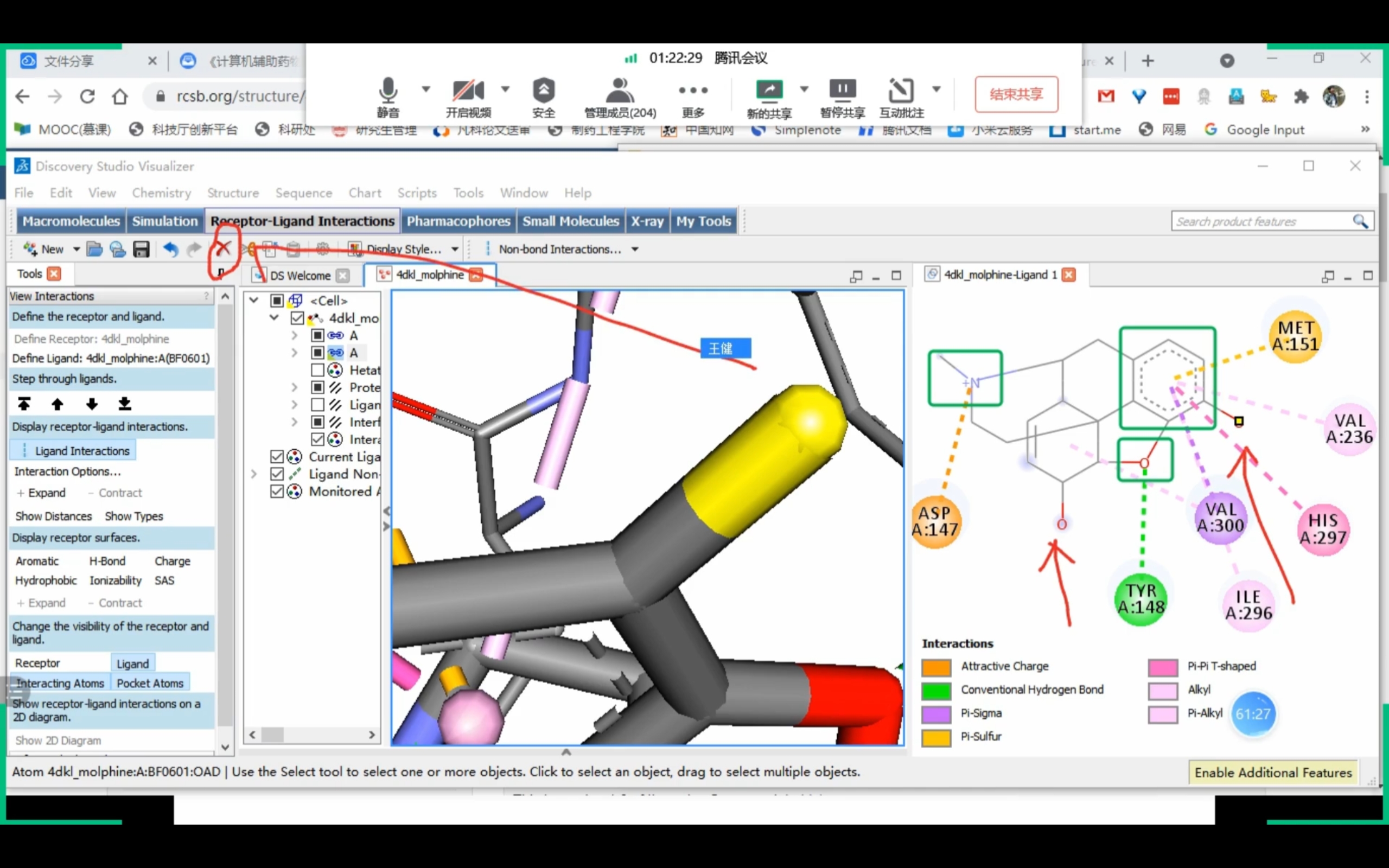
药效团确认
- 分析同一蛋白的多个晶体结构
- Protein Contacts Atlas 获取关键氨基酸
- 查阅文献中已验证药效团
- 综合by ligand与by protein结果
- 分子动力学模拟 获取关键氨基酸 (e.g. Schrödinger)
Screening
- ZINCPharmer: 工具某个PDB的ligand位,在线screening ZINC化合物数据库
- Pharmit:选择数据库中PDB与其Ligand,得到药效团3D模型(可以on-off相关特征);随后可以在线Screening:Search MolPort
- LigandScout: 设置有ZINC数据库(Screening Settings -- Database to screen),只要读取蛋白晶体结构,选中ZINC小分子,会自动生成药效团3D展示(2D图也可)。它有Ligand-Based和Structure-Based模式
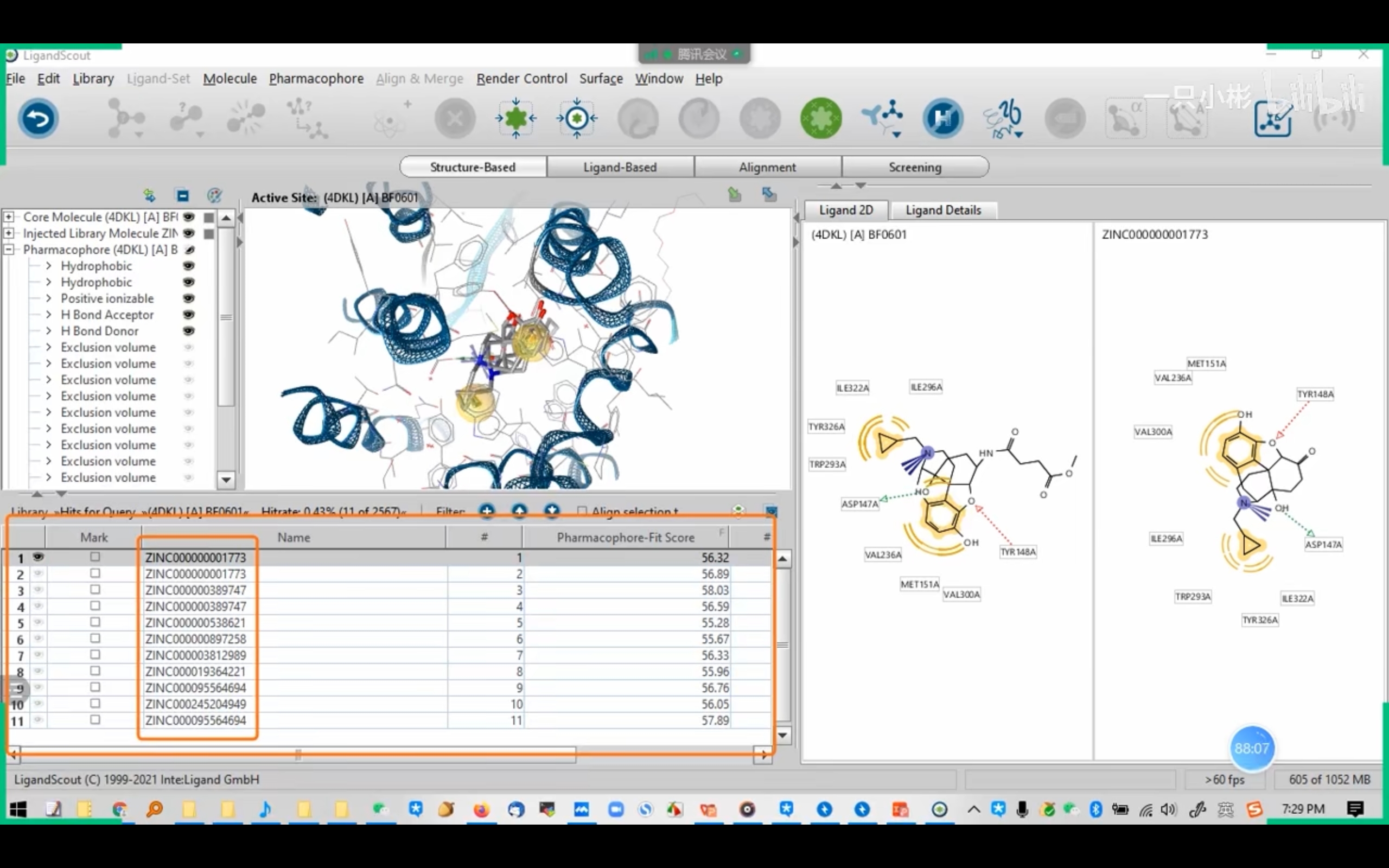 !
!
基于分子对接
将小分子放置于受体蛋白的活性位点处,寻找合适的取向和构象,使得ligand与receptor的形状和结合自由能最佳。
- 刚性/柔性对接等,TODO:待尝试Gromacs。
- ZDOCK:在线对接2个pdb蛋白
- iGEMDOCK
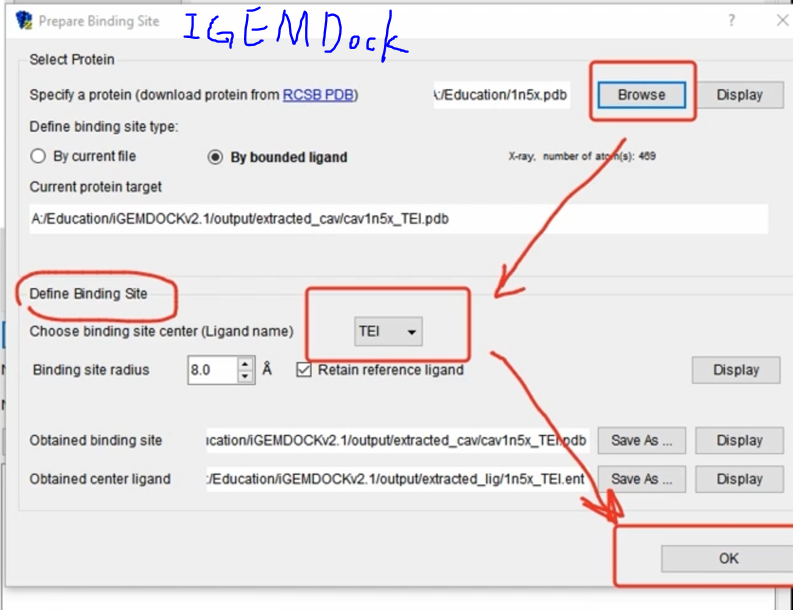 !
! - Discovery Studio 内置了AutoDock Vina等
- Schrödinger部分报告(来自课程截图)
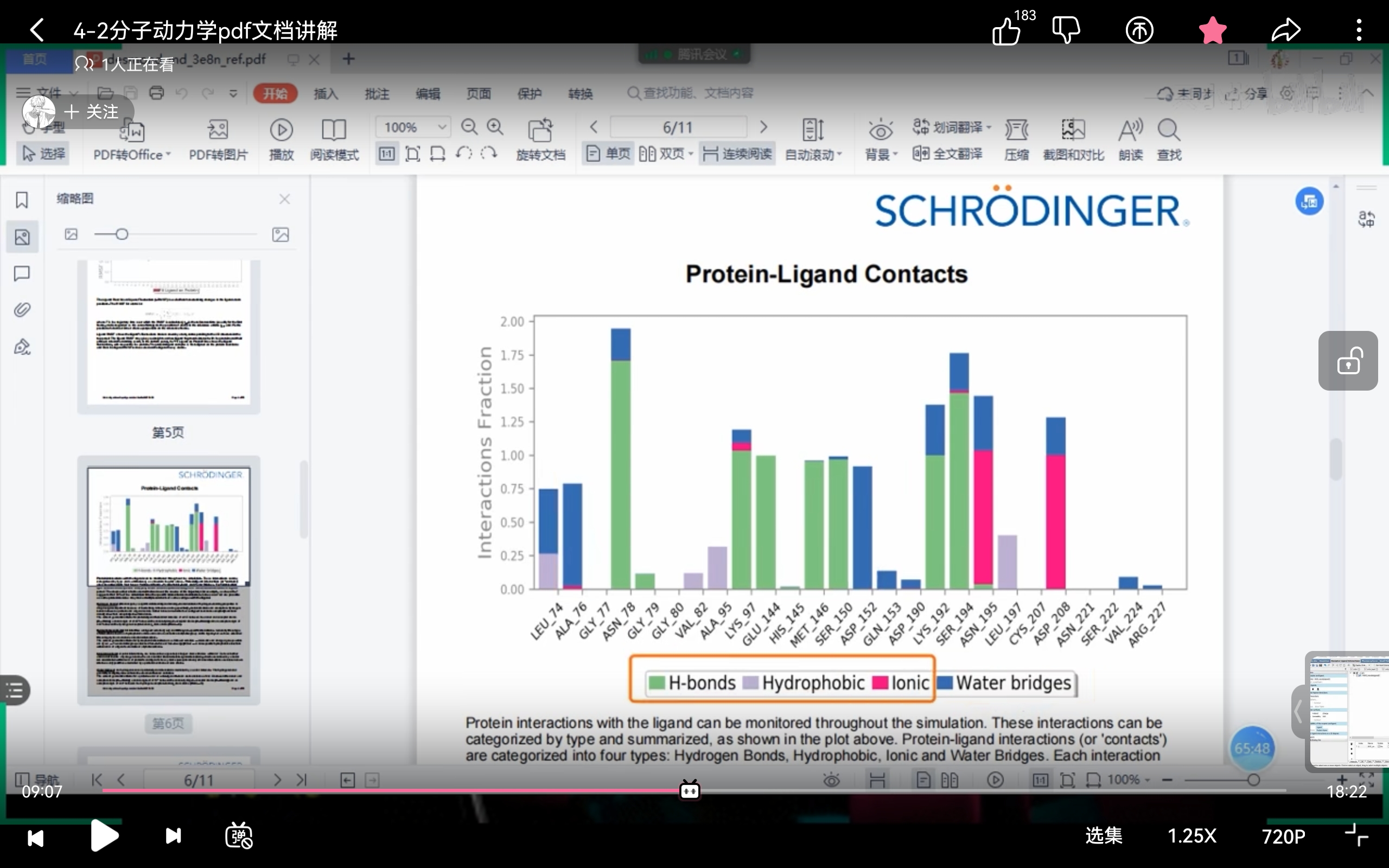 !
!
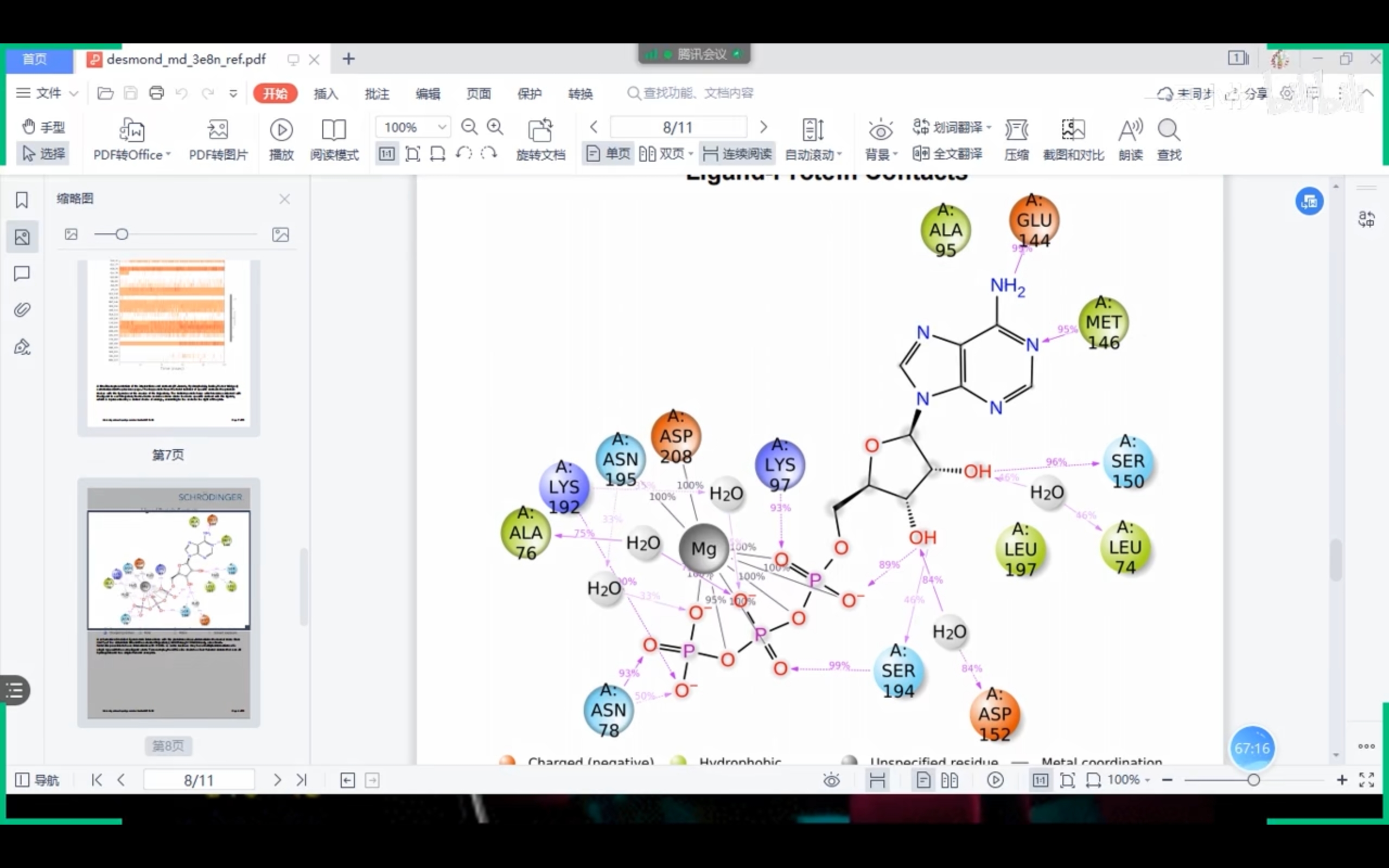 !
!
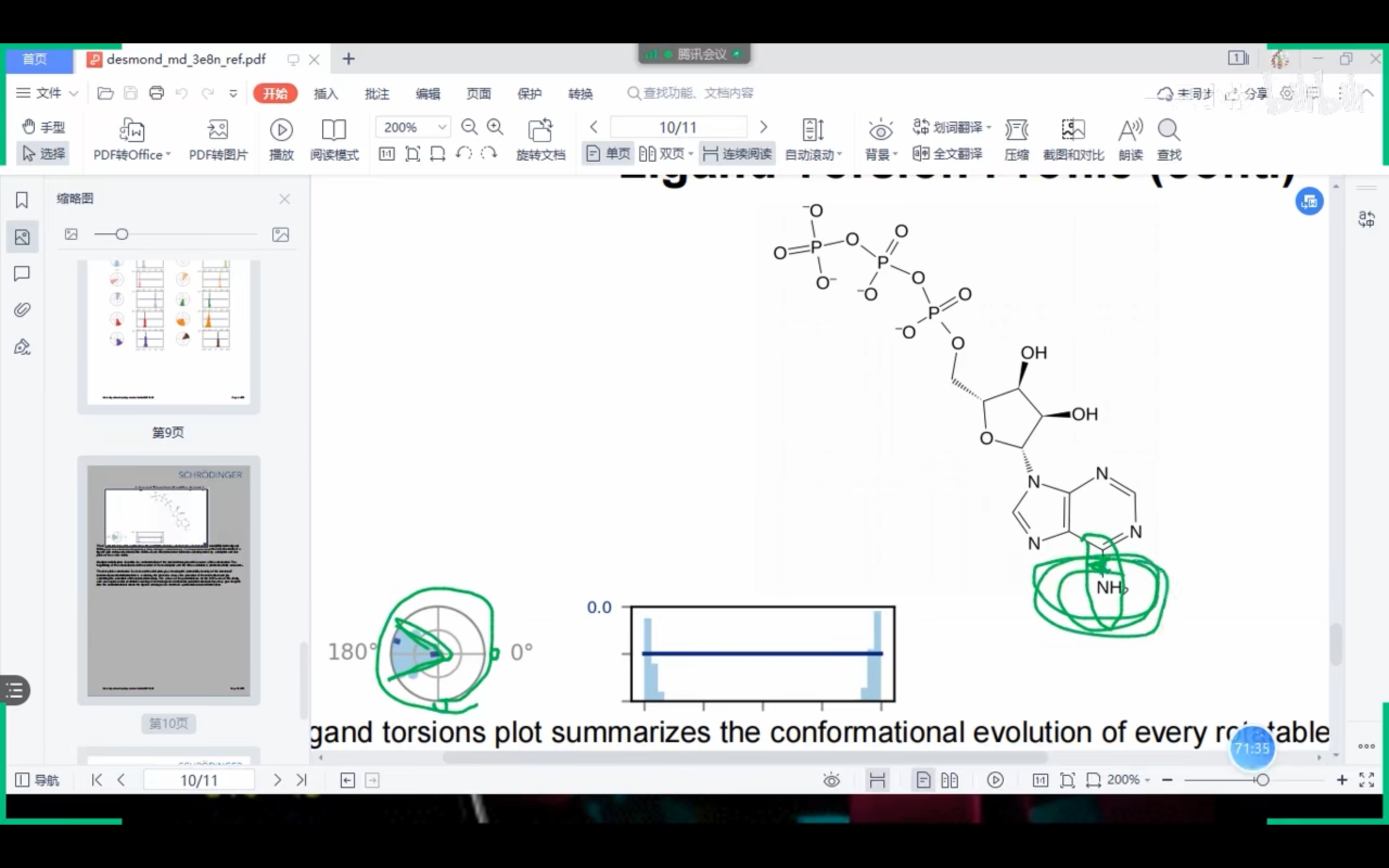 !
!
 !
!
优化先导化合物
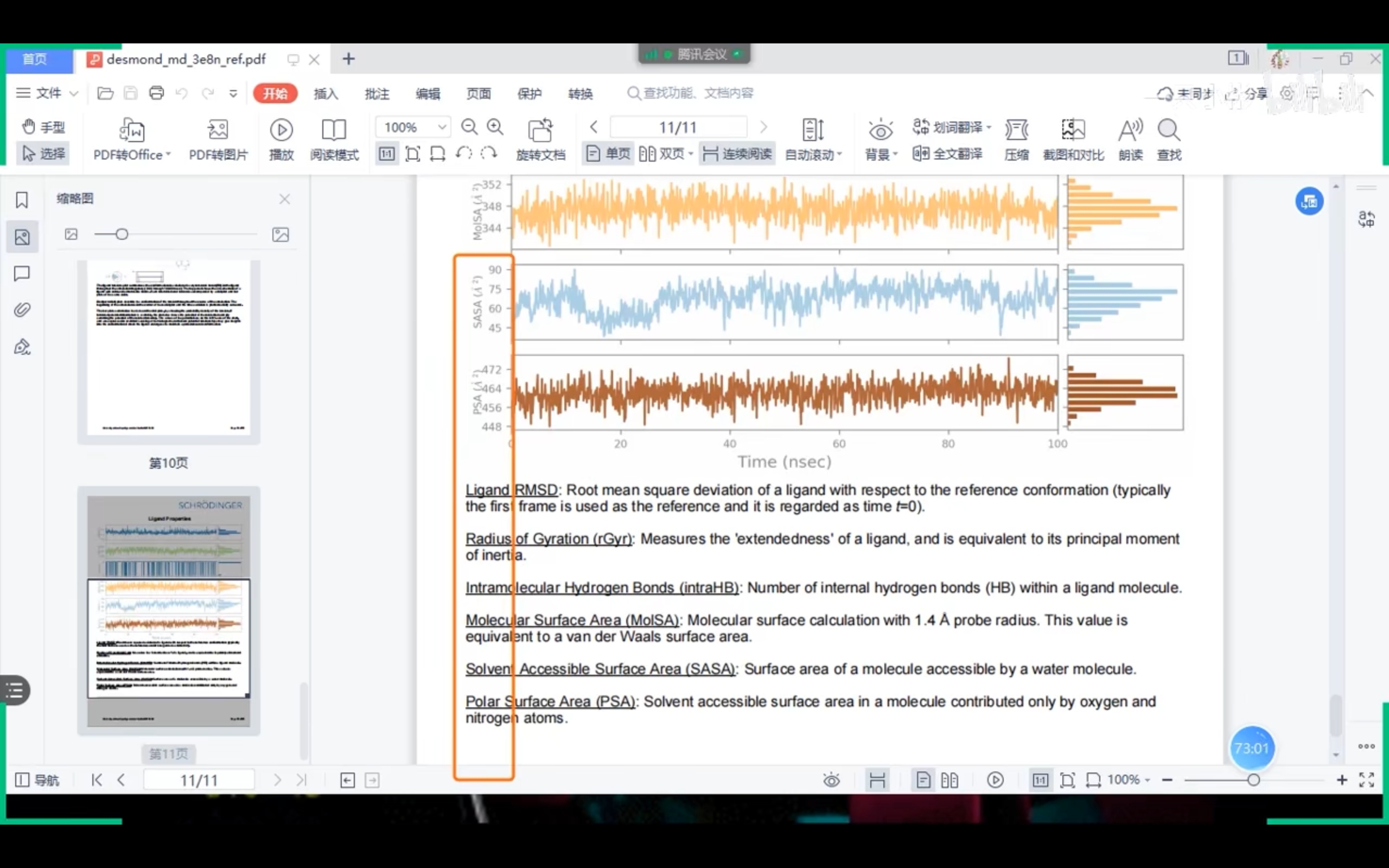
修饰先导化合物,得到其衍生物;尽量降低衍生物与Target蛋白的结合自由能(e.g. -10 kcal/mol TO -16 kcal/mol)。
- Discovery Studio中修改小分子,或者修改1D格式,待学习
药性评估
药物代谢动力学:ADME - 吸收,分布,代谢,排出,毒性(e.g. hERG毒性)
类药性:Lipinski(rule of five,基于2287个分子,便捷但粗放,Drug likeness DB 中 76.36%符合),Ghose,Oprea
 !
!
- 在线工具-swissadme:http://www.swissadme.ch/
- 在线工具-molsoft:https://molsoft.com/mprop
- 在线工具-pkcsm:https://biosig.lab.uq.edu.au/pkcsm/
- 在线工具-ADMETlab:https://admetmesh.scbdd.com/
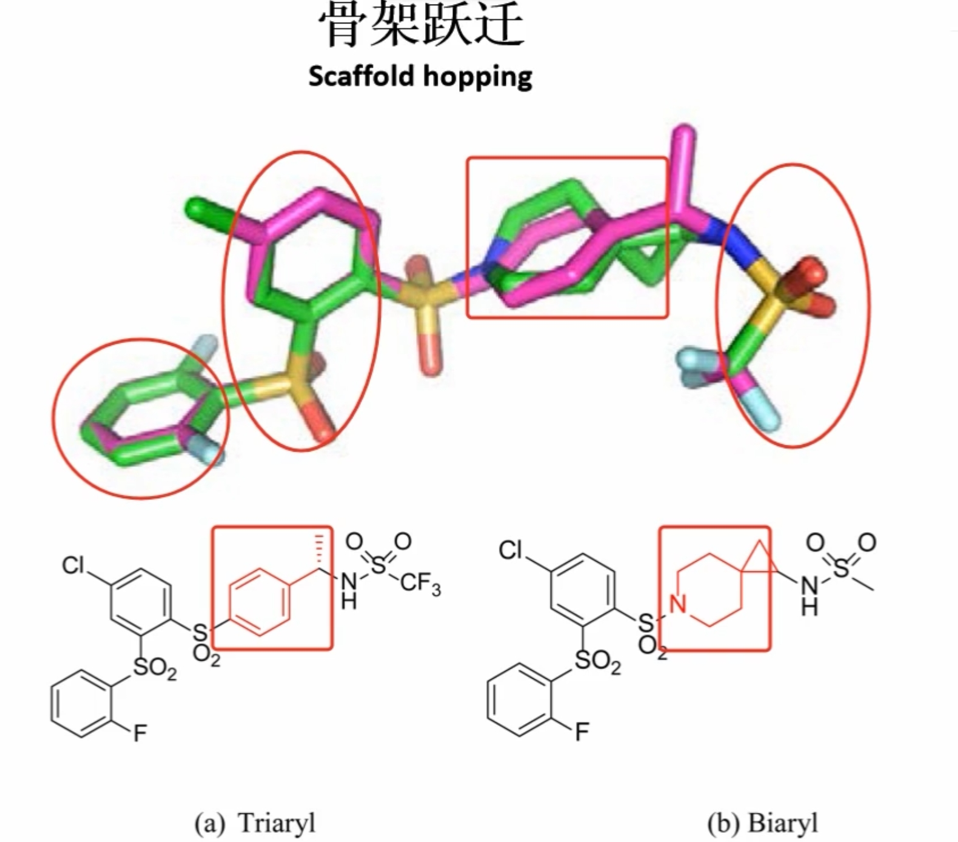
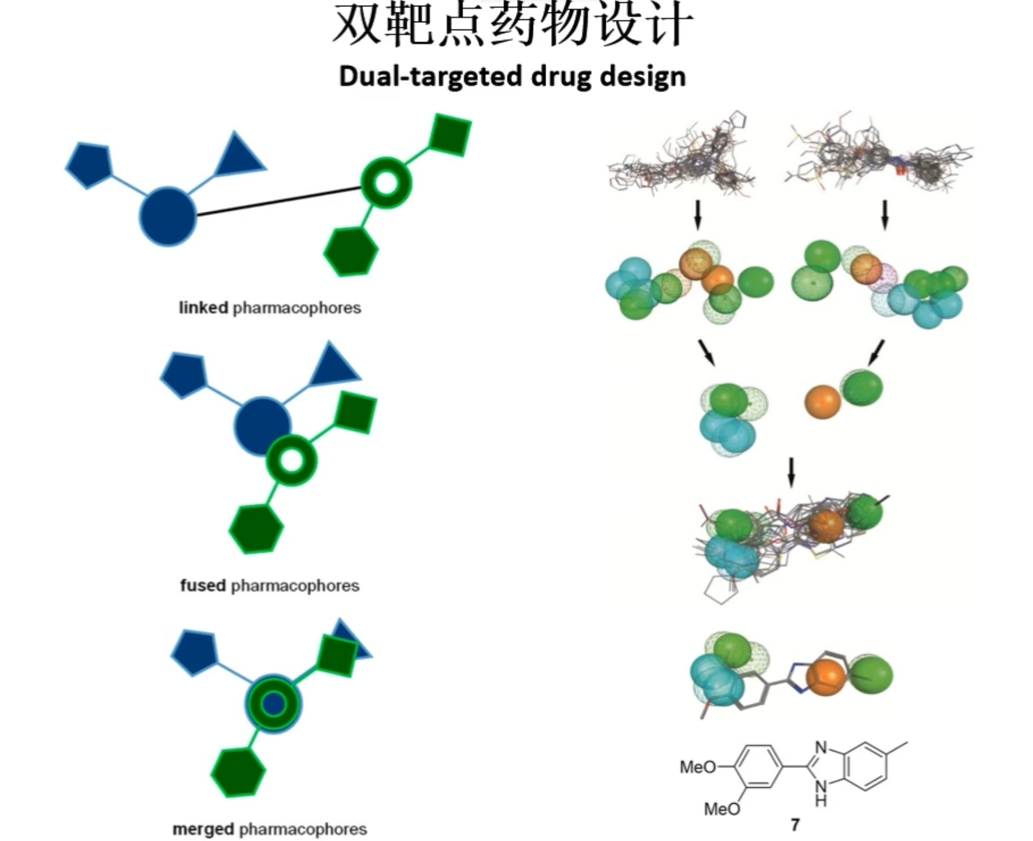
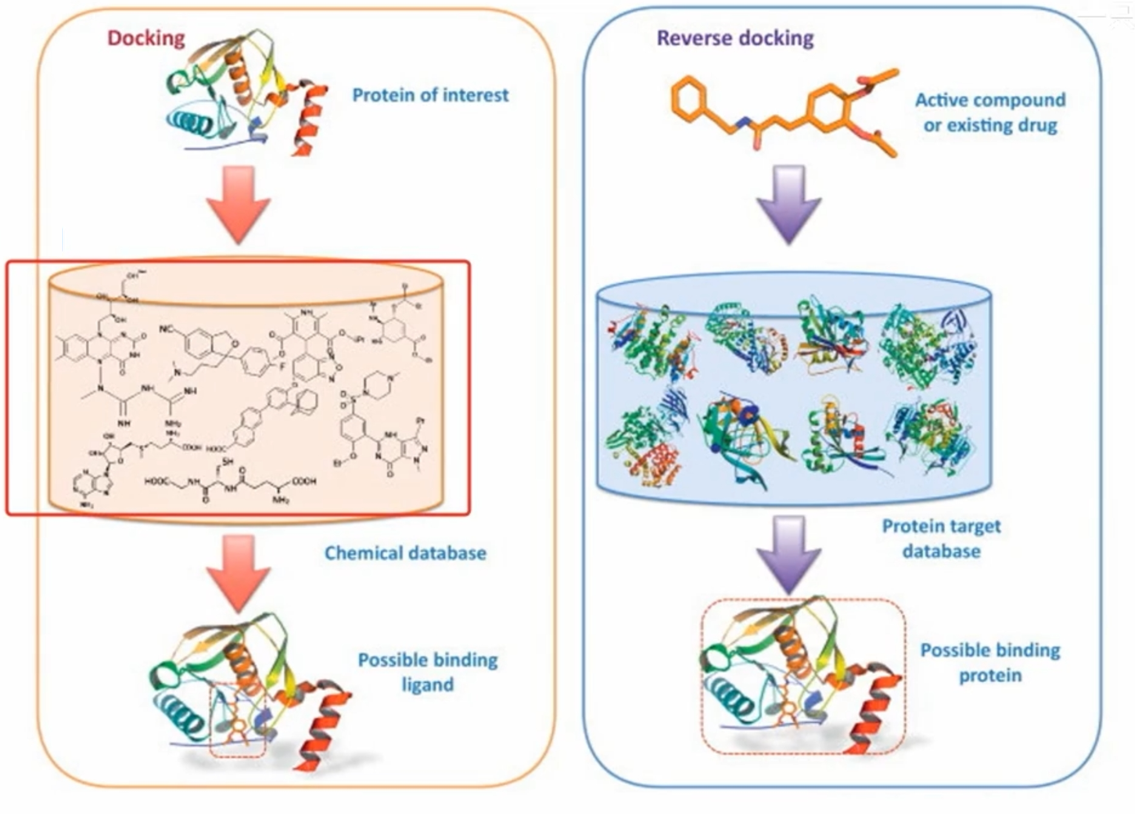
全新药物设计
 !
!
 !
!
 !
!
其它相关概念

参考&其余
Discovery Studio: https://zhuanlan.zhihu.com/p/135307440
LigPlot: https://zhuanlan.zhihu.com/p/470635337
Ligplot & Poseview: https://zhuanlan.zhihu.com/p/366602798
Schrödinger: https://zhuanlan.zhihu.com/p/401697711
Maestro: https://zhuanlan.zhihu.com/p/401872578
pkCSM: https://zhuanlan.zhihu.com/p/588494965
Materials Studio: https://zhuanlan.zhihu.com/p/340196124
配体结构修正: https://zhuanlan.zhihu.com/p/489114015
分子对接: https://blog.csdn.net/weixin_42655515/article/details/113706516
RDKit: https://zhuanlan.zhihu.com/p/82497166