FBA
得到高质量的MAGs后,鉴定出所有的酶和代谢反应(KEGG/MetaCyc/自动化工具),即得到GEMs(Genome-scale metabolic model)。(有点像MetaGEM workflow,它加了一步用 Smetana 进行 cross-feeding 模拟,用 memote 生成模型报告)
{kind=link}
如何评估GEM建造方法?可用使用包含表型信息的数据库(e.g.xx功能的酶活性),用方法重建GEM、预测表型、与原数据库信息对比
得到GEMs (SBML格式 --> 超图) 后,即可开始 Flux Balance Analysis(FBA)--- What is flux balance analysis? - PDF
FBA不修改代谢模型,它只是评估不同Flux分配方案带来的产出
Metabolic Model ∈ Virtual Cell
代谢模型是由一系列代谢反应组成的 Graph(Node=Metabolites,Edge=Reactions),边权固定为化学系数
化学反应权重固定,但反应速率可以不同;Flux/通量指代谢反应在单位时间、单位细胞质量下的转化速率:e.g. 代谢反应A ⇋ B的net_Flux = forward - reverse,可能在某种情况更多A → B、另一种情况下更多A ← B
数值上,负数表示消耗、正数表示产出,e.g. Reaction: A + 2B -> 3C 的反应系数可写作 {'A': -1, 'B': -2, 'C': +3}
代谢模型 & 一些常见的网络结构:
假设有一个Cell
─────────
Outside ┼→A → B ┼→ 2个exchange反应、1个internal反应
─────────
┌→ v2 (分流1)
v1 → A ┼→ v3 (分流2) 分流点
└→ v4 (分流3)
v1 → ↘
v2 → → B → v5 汇聚点
v3 → ↗
A → B → C → D Loop
↑ ↓
└───────────┘
┌→ R1 → R2 ┐
Start → → → → End 并行
└→ R3 → R4 ┘
A ⇌ B 可逆,flux可正可负
代谢模型重建 (基因注释EC/KO → 反应 → 代谢网络 → gap_filling / 排除假阳性、不重要的反应 ) 可以参考这篇论文。对于单个微生物物种,也可以基于基因组注释信息、从物种完整的GEM/'Universal Model'中进行删减(BiGG Models,包含物种中大量经过手动验证的高质量反应),自动化工具carveme即是如此(其它:培养基成分约束、合成辅因子/自身脂质的约束、模型简洁/最小化反应数量)
'Bottom-Up': 根据基因注释,以环境上下文的需求(以及表型)加入反应 AGORA2
'Top-Down': 根据基因注释,从'Universal Model'中进行删减 carveme、Gapseq
Gap Filling 指添加缺失反应(约束:基因组证据和生化合理性),使模型具备预期功能/使目标通路连通(因为MAGs组装并非完美)。有多种方式:从生化反应数据库/相近GEMs中补足,比对其它组学注释,基于网络的拓扑特征 GNN hyperlink prediction
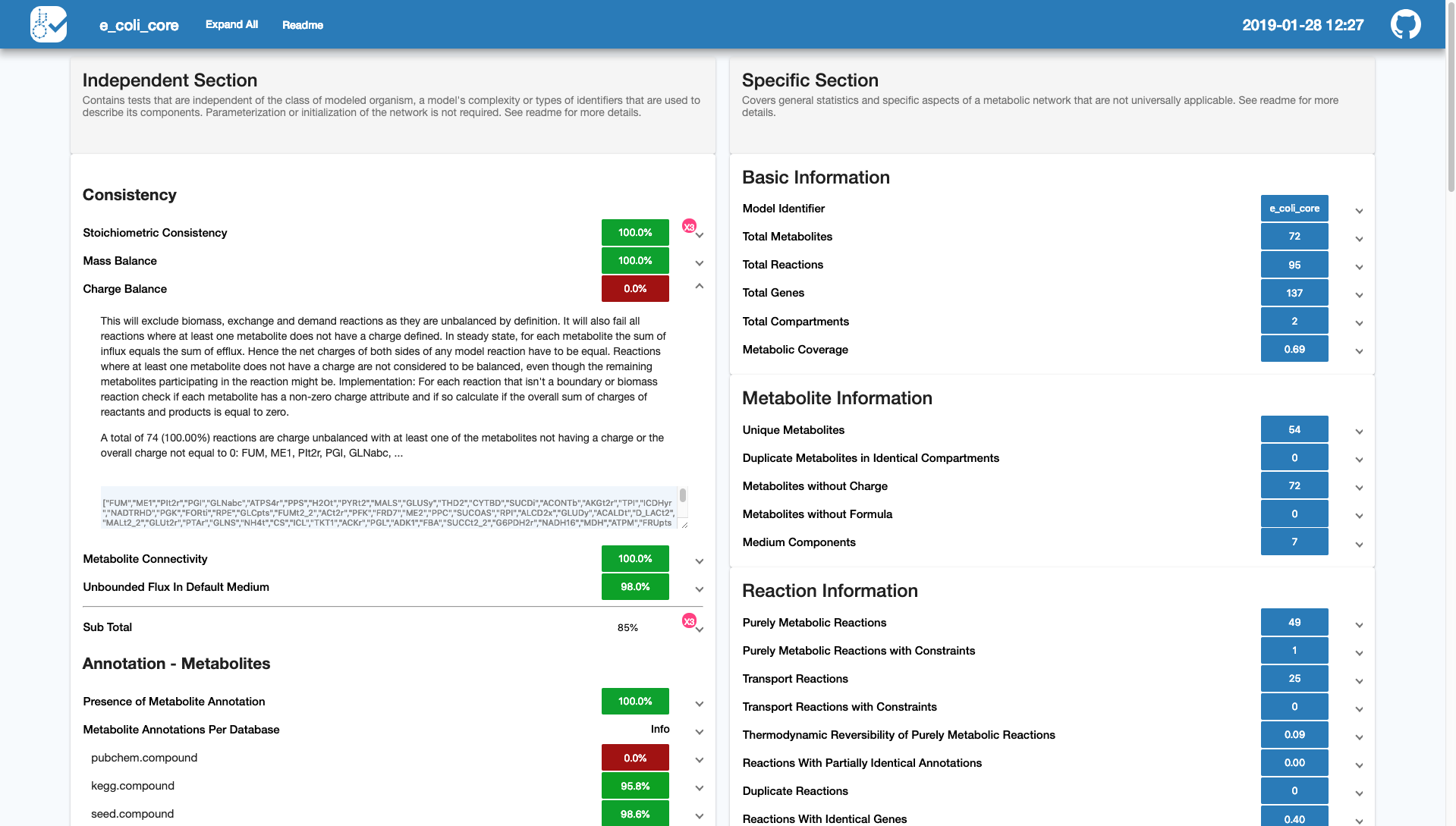
较新的GEM框架会加入生理/热力学约束(i.e.可逆/单向),e.g.GECKO 3.0/sMOMENT 构建酶约束模型(ecModel -- Flux受限于:酶活力kcat、浓度)
对于以上话题,2025年综述总结了ML如何辅助构建GEM 《机器学习驱动的基因组规模代谢模型构建与优化 李斐然等》
以上基于基因注释的是初代GSMM建模方法 单个细胞 / 隔间_互作: A-env-B / 物种泛基因组: 可变/核心
第二代建模,可以整合多组学约束:RNA/蛋白 -> 真正被表达的酶的量 -> 通路删减/约束反应上限(×酶的表达系数) GIM3E,代谢组 -> 代谢物浓度 = 反应热力学可行性(方向) mCADRE,通量组数据(如¹³C标记通量) -> 直接校准通量分布;可以整合其它生物网络,模拟不同层次的生理:GSMM + GRN + STN_信号传导网络
关于虚拟细胞的概念其实很早就有,由相对独立的功能子系统组成、使用相应的扰动实验数据训练(子)模型:
GSMM_代谢网络
GRN_基因调控网络: TF -> Target Gene
STN_信号转导网络: 受体激活、激酶级联反应、第二信使
蛋白质合成、翻译、折叠、修饰、蛋白酶体降解
膜运输、细胞器间的物质交换
细胞周期与生长:DNA复制、分裂、体积增长
根据复杂度和数据可用性,选择简单的ODE方程/复杂的随机过程模型
关于单细胞大模型/Arc虚拟细胞挑战(预测基因敲除后的细胞表达谱),只是单纯的黑盒子,它的训练数据(疾病标签/某基因的扰动)并没有覆盖全部的可能,个人怀疑它的embedding能不能反应代谢扰动? Extrapolation的问题
FBA 的约束
FBA 的约束与优化目标都是线性的,且方程式系数是整数 --- 混合整数线性规划(MILP)求解, COBRApy调用optlang包提供的优化器
maximize = objective_function
subject_to = {
# 1. 质量平衡约束,必须满足!如上图,S--模型矩阵、v--Flux向量
'mass_balance': 'S·v = 0'
# 2. Flux lower/upper Bounds,e.g. 不可逆反应的Flux≥0,环境/培养基约束(设置交换反应model.exchanges的bounds)
'capacity_constraints': 'lb ≤ v ≤ ub'
}
酶生成需要能量 --> 酶尽量少 --> internal Flux 总量尽量少
parsimonious FBA (pFBA): minimize squared sum of all fluxes while maintaining the reached optimum
整数线性规划概念 - Video,The Art of Linear Programming - Video,线性规划 standard form problem,线性规划基础
FBA 模拟示例
假设有一个大肠杆菌的GEM iML1515,模拟它在葡萄糖基本培养基中的好氧生长,希望预测最生长速率
使用 cobrapy,其中 model.objective 仅针对 Reactions,若希望优化某一代谢物,只能选取与其相关的 model.exchanges/.demands/ 反应(e.g.代谢物在 rxn.metabolites 中)
设置了Bound的反应才有这三种分类: exchanges 细胞与外部环境之间的双向交换,demands 细胞内代谢物的消耗或需求,sinks 模型填充时临时提供代谢物(BIOMASS:由各种产物汇成的输出、e.g.各种AA按细胞中比例汇成‘蛋白质集’);对于反应 Exchange: co2_e ⇋ co2/Sink: glycogen_c ⇋ glycogen,注意其代谢物 compartment 的设置,_c 细胞质隔室/_e 外部隔室
总之,参考建模教程,查看 Core Data Structures - GPR Rules
import pandas as pd
import cobra
from cobra.util.solver import linear_reaction_coefficients
from cobra.flux_analysis import flux_variability_analysis
## help(model) to see its attributes, like: .reactions .metabolites .genes
model = cobra.io.load_model('iML1515') # if web fails, use .read_sbml_model('local.xml')
## set _e intakes closed by default (-intake, +output) mmol/gDW/h
for reaction in model.exchanges:
if reaction.id not in
reaction.bounds = (0, 1000)
## set medium bounds
medium = { rxn_id:(-1000, 1000) for rxn_id in ['EX_nh4_e','EX_pi_e','EX_so4_e','EX_k_e','EX_na_e','EX_fe2_e','EX_mg2_e','EX_cl_e','EX_ca2_e','EX_cobalt2_e','EX_cu2_e','EX_mn2_e','EX_zn2_e','EX_mobd_e']}
medium.update({
'EX_glc__D_e': (-10, 1000), # Glucose
'EX_o2_e': (-20, 1000), # Oxygen, aerobic
})
for rxn_id, bounds in medium.items():
try:
reaction = model.reactions.get_by_id(rxn_id)
reaction.bounds = bounds
except:
print(f"reaction {rxn_id} not in the model")
## run FBA
model.objective = { # print(linear_reaction_coefficients(model))
model.reactions.BIOMASS_Ec_iML1515_core_75p37M: 1.0, # default obj
model.reactions.EX_ac_e: 0.5
}
assert model.reactions.get_by_id("BIOMASS_Ec_iML1515_core_75p37M").upper_bound == 1000
assert model.reactions.get_by_id("EX_ac_e").upper_bound == 1000
solution = model.optimize('maximize') ## {None, 'maximize' 'minimize'}
print(f"{solution.fluxes['EX_glc__D_e']:.4f} mmol/gDW/h") ## see each reactions
print(model.summary()) ## fva=0.95
flux_variability_analysis(model, model.reactions[:10], loopless=True) ## FVA -- finds the ranges of each metabolic flux
总之,设计不同的优化目标(e.g. 多产物合成=sum,碳利用效率=生长-葡萄糖消耗,...),计算理论得率,权衡不同通路的重要性/模拟不同环境下的代谢结果/模拟基因敲除后的结果(对包含这个基因的反应/这个基因.knock_out(),cobrapy提供单次敲除1-2个基因的批量模拟single_gene_deletion(model)),或筛查添加哪些反应能使模型变得可行 gapfill(failed_model, pan_model, demand_reactions=False, iterations=4)
如果最优解的一致性不佳,可能存在 blocked reactions,用 cobra.flux_analysis.find_blocked_reactions(model) 查看
此外,如果认为一些通量异常的高、或FVA范围异常大,需要考虑是否是loop造成的(对比Loopless模式的结果)
除了最优解,flux sampling (MCMC:Hit-and-Run) s = sample(model, 100) 可以探索(符合约束条件的)稳态下所有可能的Flux分布;FVA则是计算每个反应通量的极端取值范围
Flux coupling analysis (FCA) 则意在发现通路间的耦合:最大/最小化某个反应的通路、查看其它反应的变化
完全耦合:v₁ = k × v₂(k为常数) e.g.糖酵解中的连续步骤
方向耦合:如果v₂ > 0,则v₁ > 0;如果v₂ < 0,则v₁ < 0 e.g.一个反应为另一个反应提供必需底物
部分耦合:如果v₁ ≠ 0,则v₂ ≠ 0 e.g.两个反应共享共同的代谢物池(如,辅因子NADH)
Flux-sum coupling --> 代谢物浓度之间的关系
bounds设置 / context-specific
如果有代谢组数据(一般是多个物种但暂且假设在一个整体循环内),可以根据其比例设置相关反应的通量约束bounds,或许也可以写一个壳子来优化这些约束(e.g.gradient)/其它Graph模拟。FBA模型本质上只是在拟合不同通路的权重,只能依据生产/消耗反应的通量来间接影响代谢物的预测浓度('flux-sum')、模型中最多设置 model.add_boundary(model.metabolites.xx_c, type='demand')
如果有转录组/蛋白组,则知道了酶的丰度,可以基于酶丰度、假设其参与的反应的流量:对于各反应的 GPR Rules Enzyme_A or Enzyme_B(sum_abdance), Enzyme_A and Enzyme_B(min_abdance)
获得各反应假定的流量后,选取其中高表达者作为 'core reaction' 投入 FASTCORE,得到 context-specific GEM
(也可以:使用 StanDep 从转录组获取 'core')
把 宿主模型、微生物模型、core 都喂给 FASTCORE,那就可以建立 context-specific、Host-Microb metamodel,参考 '微生物代谢-宿主生理'论文笔记
代谢模型 + 群落模拟
gapseq 也是基于通用反应数据库的序列比对注释(e.g.NR->MetaCyc)从通用模板中建立 Draft Model、进行化学计量和可逆性的修正(ATP能量耦合、NADH辅因子平衡),Gapfilling时也考虑用户定义的培养基(必需的合成途径完整 - 如氨基酸、核苷酸、脂质前体) --- 注释搜索范围更广、手动整合生化规则,但基于序列同源性的比对忽略了蛋白质结构域、对酶功能的预测不准确:gapseq 在预测阳性表型(功能存在)方面比其他方法表现更好,但在预测阴性表型(功能不存在)方面却与其他方法不相上下
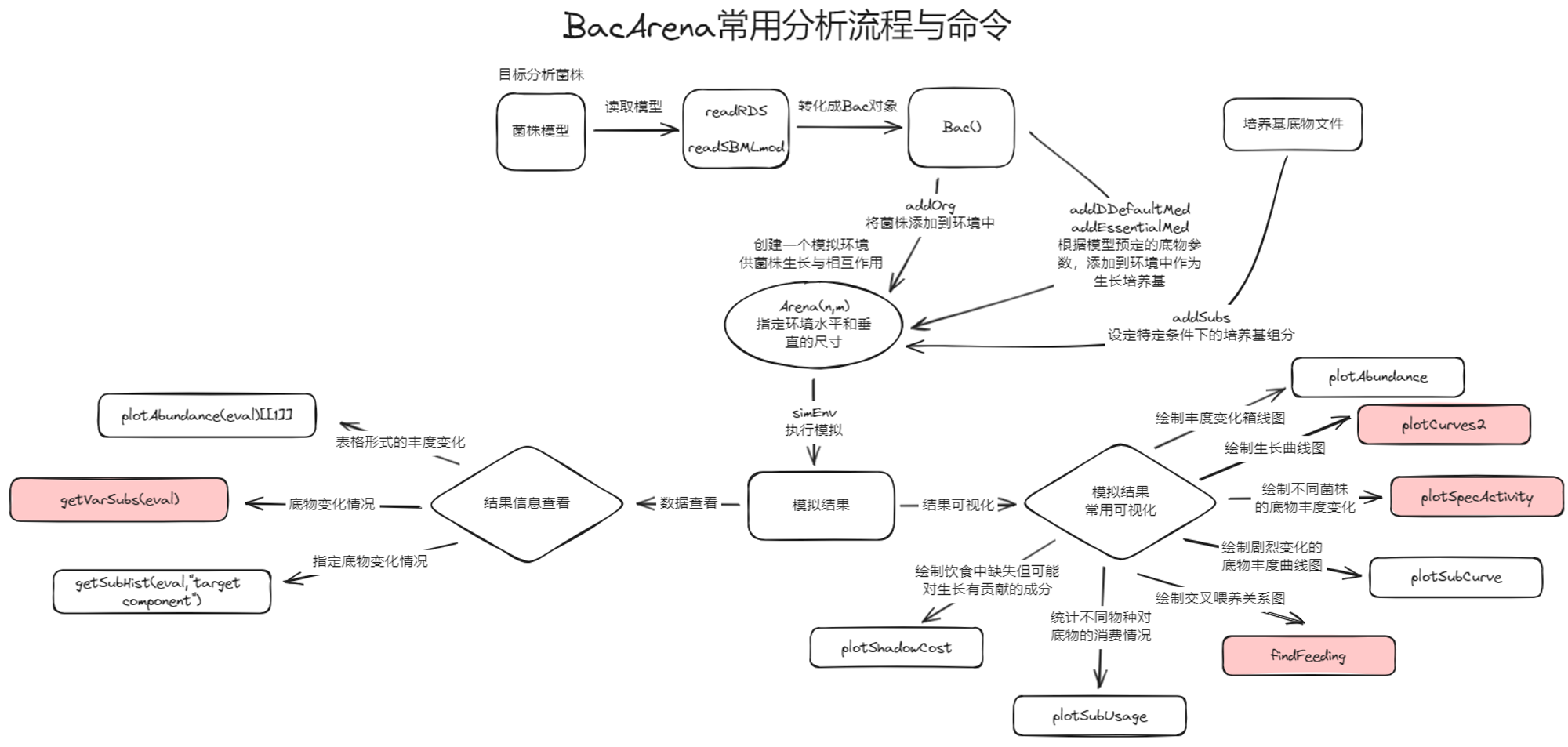
BacArena 一种微生物的代谢产物成为另一种微生物生长所需营养物质 (Cross-feeding),可以在网格世界中(Arena)模拟这个过程
1. 将n个物种模型随机放入Grid中
2. 定义初始的扩散基质(如葡萄糖、氧气--记得移除Cross-Feeding的代谢物以突显效果)及其在网格中的初始浓度、扩散系数
3. 模拟T个timesteps
- dFBA:依据当前环境中代谢物浓度,得到最优生长速率μ的Flux分布
- 按当前FBA结果更新环境
- 生物量达到初始值两倍后细胞分裂(减半、扩散到相邻格子)
Virtual Colon 是 BacArena 的扩展,提供宿主结肠环境的设计:查看原文 TABLE 1
blood | cell | inner mucus | outer mucus | lumen
有五个区隔,细菌只能生活在粘液层和空腔,粘液层越厚、分子扩散系数越小
网格尺度、体积则参考解剖学
营养基为标准膳食、另加入血液代谢物,仅匹配细菌的交换反应
宿主模型则参考小鼠单细胞转录组的结肠细胞!! -- 宿主细胞固定,细菌细胞游走
The virtual host cells inherited the default human class settings of BacArena 指什么?
一部分宿主细胞与放入的特定细菌物种交互,另一部分与默认肠道菌群的简化模型交互?