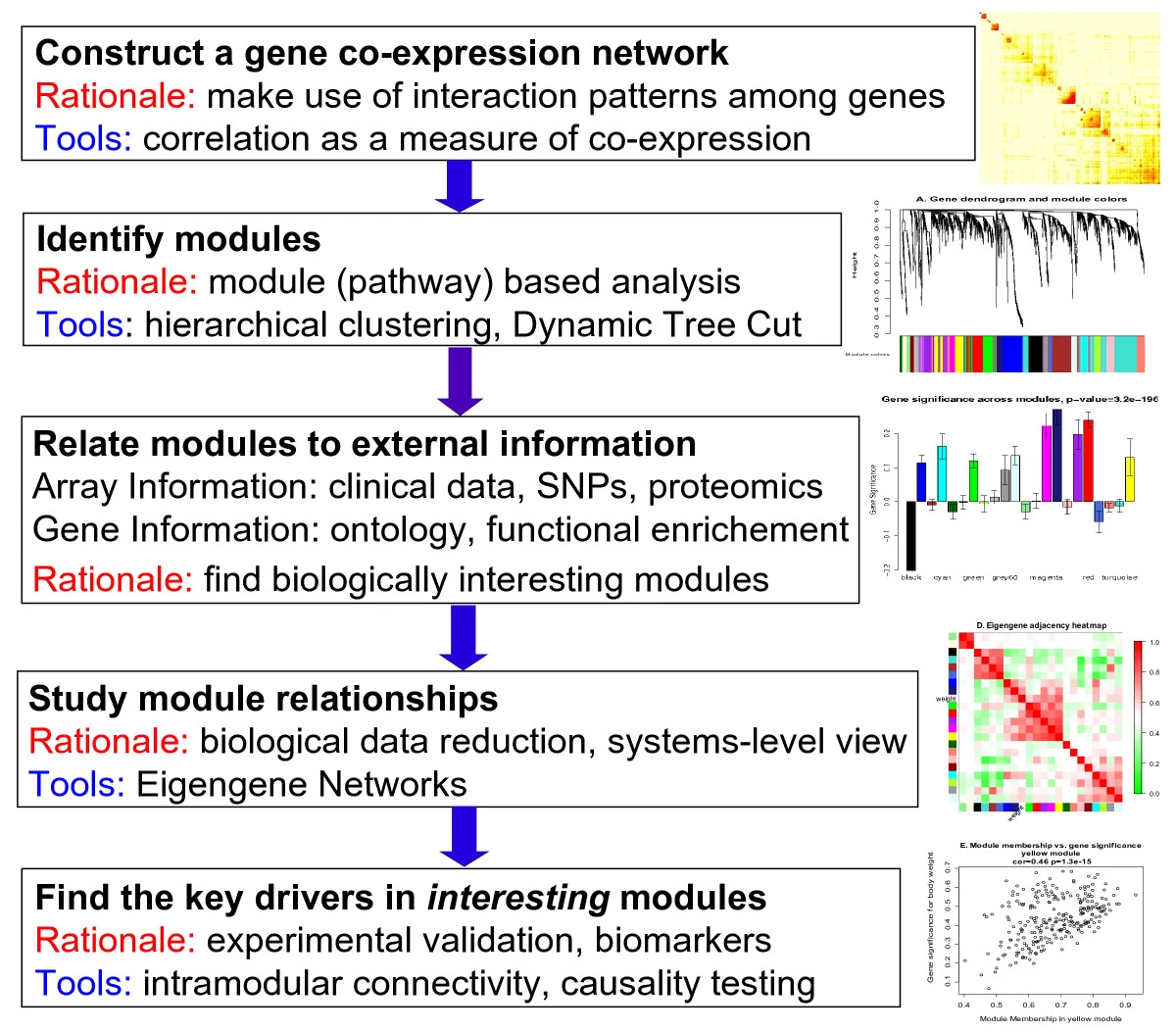
WGCNA
RNA项目的WGCNA: 基因模块是否在不同分组间有差异?关注一组共表达的基因(i.e.模块)而不是单个差异基因
R
BiocManager::install("WGCNA") 进行安装
教程:Network analysis with WGCNA,中文讲解
输入:表达矩阵(行=样本,列=基因),需要知晓样本的分组;一般需要>15样本,且建议输入全部基因、不要仅选择差异基因
## wget ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE61nnn/GSE61333/suppl/GSE61333_ligule_count.txt.gz
## gunzip GSE61333_ligule_count.txt.gz
library(tidyverse)
library(magrittr)
library(DESeq2)
library(WGCNA)
## Load matrix
load('input_mat.rdata') ## normalized input matrix, we can check rna-counts of each sample via violin_plots
load('meta_df.rdata') ## group_info
########## WGCNA ##########
## 1. Select soft-thresholding powers for network building
## sft$fitIndices: Power, SFT.R.sq, slope, truncated.R.sq, mean.k., median.k., max.k
## X-axis: Power (=sft$fitIndices[, 1])
## Y-axis: mean.k. or -1*SFT.R.sq*slope (sft$powerEstimate decided upon this value)
powers = c(c(1:10), seq(from = 12, to = 16, by = 2))
sft = pickSoftThreshold(input_mat, powerVector = powers,verbose = 5)
## 2. Build a complete network
net <- blockwiseModules(input_mat,power = sft$powerEstimate,networkType = "signed")
## 3. Plot modules, ...
moduleColors <- labels2colors(net$colors)
plotDendroAndColors(
net$dendrograms[[1]],
moduleColors[net$blockGenes[[1]]],
"Module colors",
dendroLabels = FALSE,
hang = 0.03,
addGuide = TRUE,
guideHang = 0.05
)
## 4. Genes in each modules: gene_id, colors
module_df <- data.frame(
gene_id = names(net$colors),
colors = moduleColors ## labels2colors(netwk$colors)
)
## 5. Module-trait(Group) relationship: based on PC1 of each samples in each module (the so-called eigengenes)
sample_num <- nrow(value)
value <- moduleEigengenes(input_mat, moduleColors)$eigengenes
trait <- matrix(rnorm(sample_num) , nrow =sample_num)
p_val <- cor(value, trait, use = "p")
p_val_adj <- corPvalueStudent(p_val, sample_num)
# > p_val_adj
# [,1]
# MEblack 0.261063268
# MEblue 0.375112835
# MEbrown 0.955115638
# MEcyan 0.571728398
- 其中,灰色区域表示无法聚类到模块中的基因,如果占比过大,说明总体共表达趋势不明显,需要调整预处理方法/过滤基因
- 如果出现
Error in (new("standardGeneric", .Data = function (x, y = NULL, use = "everything",可能是包的冲突,强行令cor <- WGCNA::cor