Circos
circlize
Data:bed/Cytoband
library(circlize)
library(dplyr)
df = read.cytoband()$df
colnames(df) = c('chr','start','end','name','stain')
sector_data = as.factor(df$chr)
sector_lvls = as.factor(levels(sector_data))
x_data = runif(nrow(df))
y_data = runif(nrow(df))
xlim = c(0,1)
ylim = c(0,1)
df$GC = runif(nrow(df))
Initialize
设定坐标系, 依次设定 sector - xlim - ylim
Opt-1 Basic
可以直接输入vector,会自己计算 sectors xlim ylim;也可以输入sector_lvls xlim ylim
circos.initialize(
sectors = sector_data, # factor vector, categories
xlim = xlim,
sector.width = rep(c(0.10),length(sector_lvls)) # Width for each sector
)
Track Region显示axis (传入circos.trackPlotRegion)
circos.track(
ylim = ylim,
panel.fun = function(region, value){
circos.axis(labels.facing='reverse.clockwise')
}
)
只使用部分sectors(但还是显示了sector方框)
circos.trackPlotRegion(sectors = c('chr1','chr10'),
ylim = ylim,
panel.fun = function(region, value){
circos.axis(labels.facing='reverse.clockwise')
}
)
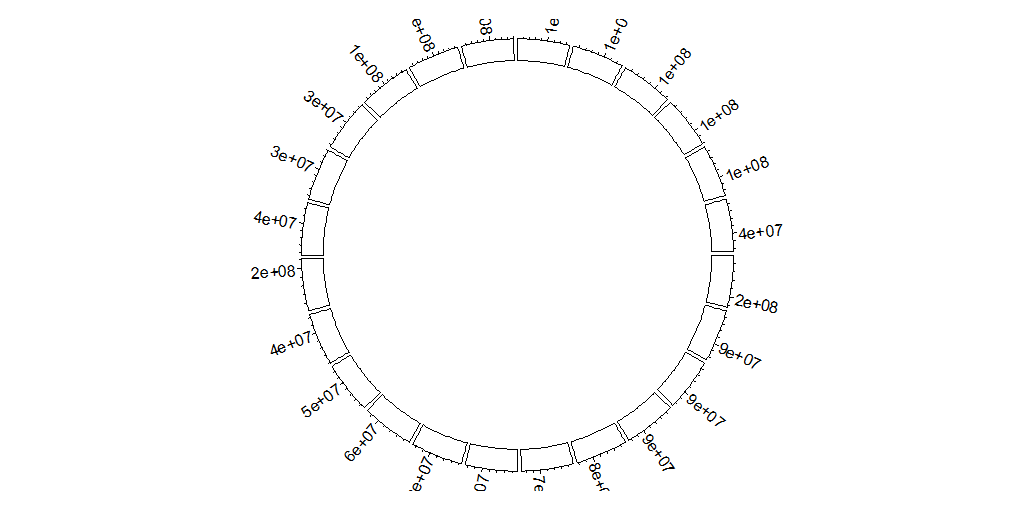
Opt-2 Genome
必须带有Cytoband信息。 本例中,每个sector的xlim通过df自动计算,故而范围决定于表中基因位置的边界;实际使用中init这一步应当输入各个chr的总长度bed_df(每一行: chr_name 0 chr_length ...)
circos.genomicInitialize(
data = df,
major.by = 10000000,
plotType = c('axis','labels','ideogram'),
tickLabelsStartFromZero =T,
axis.labels.cex = 0.5,
labels.cex = 1,
track.height = 0.10,
)

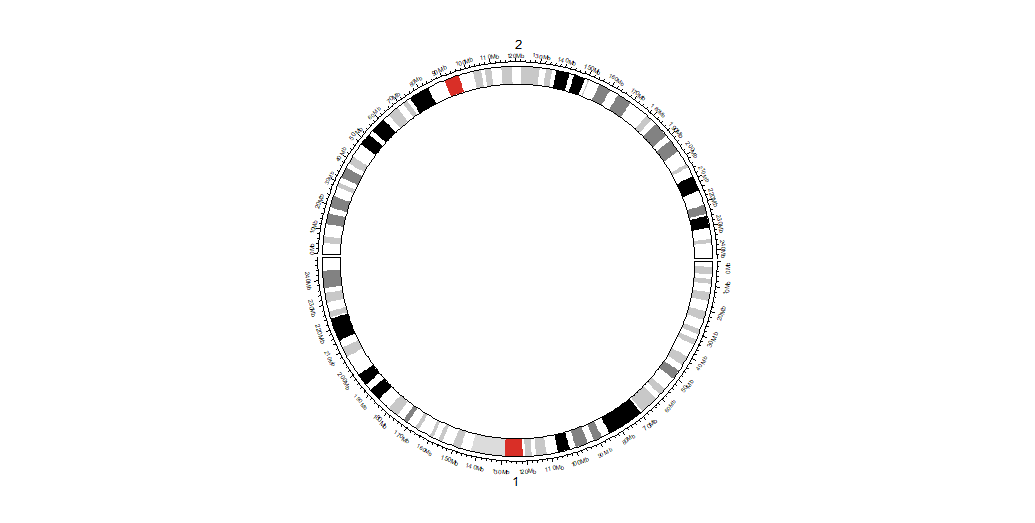
Opt-3 Genome + Ideogram
circos.initializeWithIdeogram(
cytoband = df, ## or: cytoband_file_dir
chromosome.index = c('chr1','chr2'), ## conflict with sort.chr
major.by = 10000000,
plotType = c('axis','labels','ideogram'),
track.height = 0.10,
ideogram.height = 0.08
)
## 注:可以 circos.genomicIdeogram() 多次多层
## 注:plotType可以控制是否在init时候画Ideogram
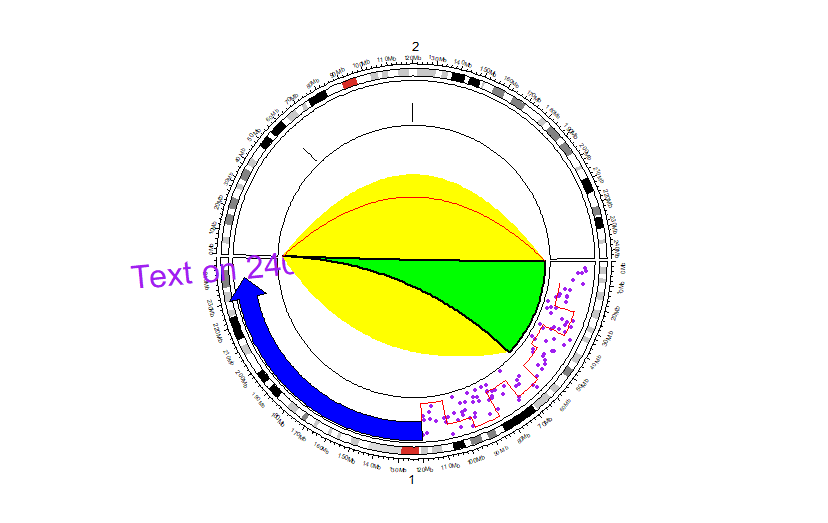
Functions
所有方程、使用方法见官方文档,或通过help(),或??circlize
Opt-1 单独使用
(指定sector.index与track.index)
circos.initializeWithIdeogram(cytoband = df,chromosome.index = c('chr1','chr2'))
circos.track(ylim = ylim)
circos.points(runif(100)*120000000, runif(100), ## X_lst y_lst
sector.index = 'chr1',
pch = 16,
cex = 0.5,
col = "purple")
circos.text(240000000, 1, ## X_lst y_lst
sector.index = 'chr1',
labels = "Text on 240Mb",
facing = "reverse.clockwise",
adj = c(0.5, 0),
cex = 2,
col = "purple")
circos.arrow(120000000,240000000,0.8, ## x_start x_end y_middle
sector.index = 'chr1',
col = "blue")
circos.lines((1:10)*12000000, runif(10),
sector.index = 'chr1',
type = "s",
col = "red")
## value(y_lst) pos(x_lst)
circos.boxplot(runif(100)/2, c(rep(60000000,50),rep(120000000,50)),
sector.index = 'chr2',
col = "red")
circos.link('chr1', c(10000, 60000000), ## From x
'chr2', c(10000, 20000), ## To x
col = 'yellow',
h = 0.2,
border = NA) # interval to interval
circos.link('chr1', c(10000, 60000000),
'chr2', 0,
col = "green",
border = "black",
lwd = 2) # point to interval
circos.link('chr1', 0, 'chr2', 0, h = 0.3 , col = 'red') # point to point
Opt-2 panel.fun + genomicTrack
查看一下传入的参数
circos.initializeWithIdeogram(cytoband = df,chromosome.index = c('chr1','chr2'))
circos.genomicTrack(df,
ylim = ylim,
panel.fun = function(region, value,...){
print('--------region--------------')
print(head(region,n=2))
print('--------value--------------')
print(head(value,n=2))
print('--------df--------------')
print(head(df,n=2))
}
)
## (region , value, ...)
## region: two-col df with start_pos and end_pos in current genomic category (e.g. chromosome)
## value : df which is derived from data but excluding the first three columns.
## Rows in value correspond to rows in region
## ... : mandatory and is used to pass internal parameters to other functions
结果
[1] "--------region--------------" ## chr1
start end
1 0 2300000
2 2300000 5400000
[1] "--------value--------------"
name stain
1 p36.33 gneg
2 p36.32 gpos25
[1] "--------df--------------"
chr start end name stain
1 chr1 0 2300000 p36.33 gneg
2 chr1 2300000 5400000 p36.32 gpos25
[1] "--------region--------------" ## chr2
start end
371 0 4400000
372 4400000 7100000
[1] "--------value--------------"
name stain
371 p25.3 gneg
372 p25.2 gpos50
[1] "--------df--------------"
chr start end name stain
1 chr1 0 2300000 p36.33 gneg
2 chr1 2300000 5400000 p36.32 gpos25
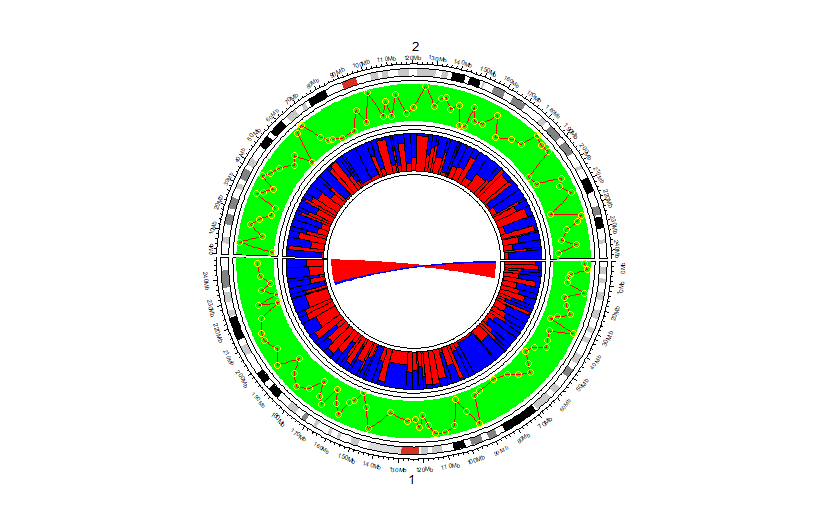
使用示例:
circos.initializeWithIdeogram(cytoband = df,chromosome.index = c('chr1','chr2'))
circos.genomicTrack(
df[c(colnames(df)[1:3],"GC")],
ylim = ylim,
panel.fun = function(region, value,...){
circos.genomicRect(region, value, col = 'green', border = NA, ...)
circos.genomicLines(region, value, col = 'red', lwd = 0.35, ...)
circos.genomicPoints(region, value, numeric.column = "GC", col = 'yellow')
}
)
circos.genomicTrack(
df[c(colnames(df)[1:3],"GC")],
ylim = ylim,
panel.fun = function(region, value,...){
circos.genomicRect(region, value, ytop.column = "GC" ,col= 'red')
circos.genomicRect(region, value, ybottom.column = "GC",col= 'blue')
}
)
bed1 = head(df %>% filter(chr == 'chr1'))
bed2 = tail(df %>% filter(chr == 'chr1'))
circos.genomicLink(bed1, bed2, col='red', border = NA)
circos.genomicLink(head(bed1,1), head(bed2,1), col='blue', border = NA)
Others
画完后不要忘记关闭
circos.clear()
还可以设置par
circos.par()
随机生成bed数据
generateRandomBed(10)
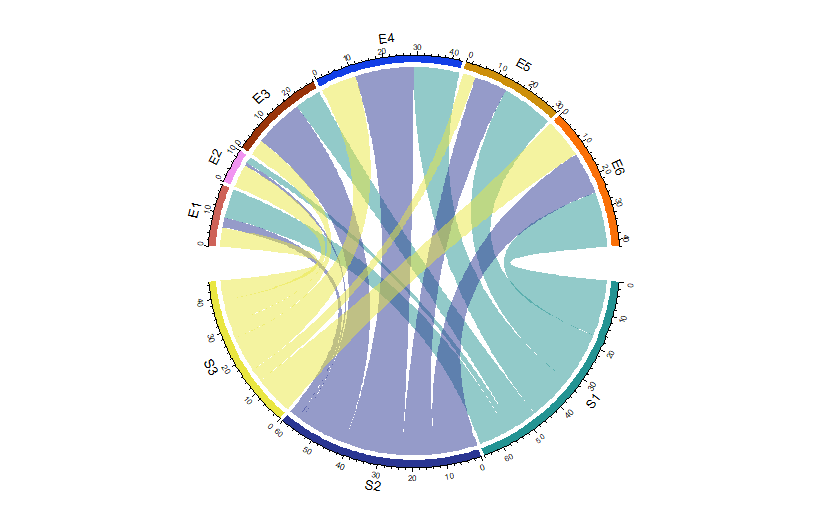
chordDiagram
包含在circlize中
Data : Adjacency Matrix
mat <- matrix(sample(18, 18), 3, 6)
rownames(mat) <- paste0("S", 1:3)
colnames(mat) <- paste0("E", 1:6)
> mat
E1 E2 E3 E4 E5 E6
S1 9 2 8 14 16 17
S2 3 1 15 18 10 13
S3 6 7 5 11 4 12
Use
chordDiagram(mat)
参考
circlize Doc: https://jokergoo.github.io/circlize/
circlize:https://www.jianshu.com/p/a87bcc1cb67b
circlize:https://mp.weixin.qq.com/s?__biz=MzIxNzc1Mzk3NQ==&mid=2247484280&idx=1&sn=fe5e0c82c123f8947b3240229ceef088&
circlize:https://zhuanlan.zhihu.com/p/376017987
chordDiagram: https://zhuanlan.zhihu.com/p/378190330
Cytoband -- Cytogenetic Bands:
https://www.ncbi.nlm.nih.gov/Class/MLACourse/Modules/Genomes/map_cytogenetic_bands.html
https://www.jianshu.com/p/9491901e3a9b
https://zhuanlan.zhihu.com/p/284010321
R: https://www.jianshu.com/p/cd957b3d6d4b
染色体显带及命名: https://zhuanlan.zhihu.com/p/284010321