All Omics Overview
- 在需要准确定量的项目如ATAC、转录组中,可以加入去除PCR重复的步骤。例如scRNA使用的UMI技术,或者通过samtools/picard去除duplicate
- RNA项目可以使用RSeQC进行测序饱和度分析,以确保有效检测
- 简化xxx:缩小测序范围,e.g. WES,RRBS
- 当部分样本直接具有关联性时,就不能简单的考虑 Sample Size。假设有一部分样本(N=2)的Correlation可以计算,则着部分样本的 Effective_Sample_Size = N/[1+corr*(N-1)]
Bulk
Basic Pipelines
| Basic Pipelines | 说明 | -- |
|---|---|---|
| Genome | 组装基因组,并且预测基因;准备上传NCBI的格式 | 其它:Haplotype-based组装;结合HiC数据的组装 |
| Transcriptomics | RNA-seq | 基因的转录水平,可变剪接 |
| Proteomics | 蛋白组 | -- |
| Metabolomics | 代谢组 | -- |
| Re-seq | 对已知基因组序列信息的个体进行测序,获取SNP、InDel、SV等信息 | 群体遗传,下游:GWAS |
Metagenomics
这里有一篇流程/工具总结
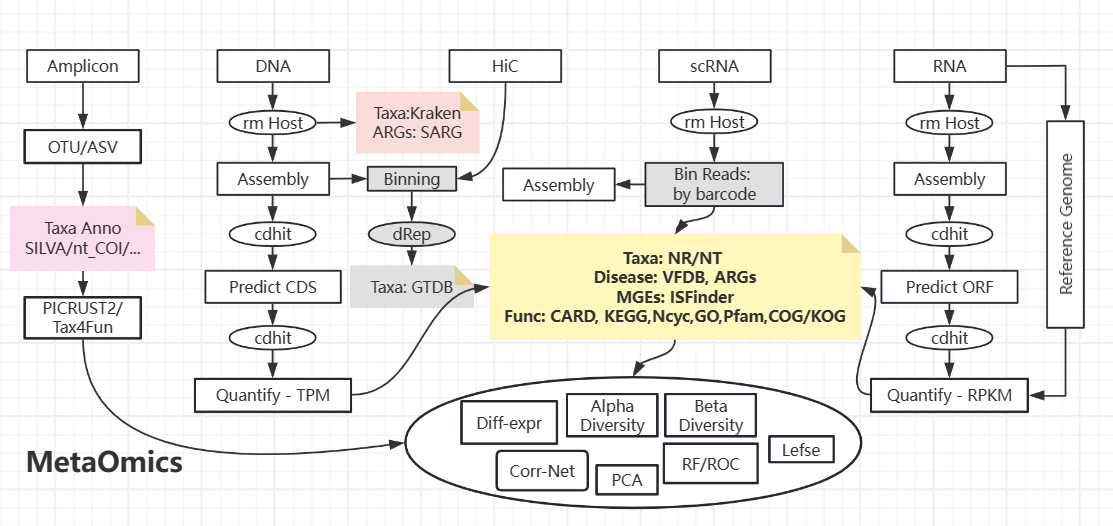
病毒由于含量少,另有工具(VirSorter/VirFinder/geNomad/...checkv)
对于Binning质量,一般使用checkm/busco进行检测 --- 注意,这俩是基于数据库的,其外的污染/缺失不能被检出
cd-hit 去除冗余序列,dRep去除冗余细胞
关注环境中物种组成,或者功能基因的比例,或者物种/功能的Network,或者进行 Flux 分析
需要注意的是,由于这类研究的结果通常使用比例的方式呈现,可能会出现一些虚假的相关性,例如下例:看起来A下降了、B/C/D的丰度上涨了,它们负相关?但其实只是A变少了,其它没变 (已经有一些定量的生态学数据库?)
AAAA BB CC DD ==> AA BB CC DD
40% 20% 20% 20% 25% 25% 25% 25%
而且微生物可能是处于动态平衡中,一次采样只是得到一个瞬时snapshot;再者MAGs本质上也是一堆相似基因组的cluster,高质量MAGs之外还有些没被归类的序列可能有功能
Epi & Regulome
表观遗传修饰表观修饰可能是影响基因表达的潜在因素,常见包括:DNA甲基化、组蛋白修饰(影响染色质的暴露)、RNA甲基化... 研究话题可能与 发育、疾病、衰老 相关,例如:什么机制调控了疾病相关xx基因的过表达?
可以用实验手段直接评估调控元件的活性,例如:MPRA(大规模、并行、基因编辑)
DNA水平上的表观修饰主要是甲基化,可通过 BS-seq 获取甲基化位点信息 / 免疫沉淀富集6mA的片段(MeDIP-seq) / 测序平台直接获取。
转录调控(DNA -> RNA Transcription)一般也需要考虑细胞内DNA的3D空间距离信息(HiC)。ATAC-seq (Tn5) 标注所有开放染色质(Peaks 信号强度 = 开放程度),这些开放染色质可能是潜在的 TF-binding site (TFBS),双峰/下凹Peak可能意味着位点被核小体/TF占据(也有可能是信号失真)。Chip-seq (超声随机断裂) 的原理是通过免疫共沉淀捕获目标蛋白、从而获得目标蛋白与DNA的互作位点,对象一般是修饰后的组蛋白/特定TF(e.g.CTCF)。CUT&Tag (Tn5 优先切割开放染色质) / CUT&RUN (MNase 对某些蛋白不敏感) 同样用于获得目标蛋白与DNA的互作,似乎更灵敏、更精细、低噪音、操作便捷,逐渐替代 Chip-seq。PRO-seq 用生物素标记原料、获得新生成的RNA,从这些被标记的RNA的位置,可知正在生成中RNA的转录起始位点(TSS)。CAGE(Cap Analysis of Gene Expression)精确捕获 mRNA 5' Cap 并测序其下游的短片段,即精确定位所有静态/稳态RNA的 TSS、CAGE tag 计数 = 启动子活性。
获知活跃的TFBS后(e.g. 与增强子活性标记 H3K27ac 共定位),如何推断其对应调控的基因?
1. 已知:增强子通常位于靶基因的±1Mb内,启动子区域(TSS ±2.5kb)的TFBS直接关联该基因
- 方法:线性距离最近的TSS(基因)
2. 3D距离最近的TSS(基因) ---- HiC 辅助
3. 假设:TF与其靶基因可能共表达
- 使用公共数据库(如GTEx、TCGA)或自有RNA-seq数据,构建共表达网络(如WGCNA)/ 筛选与TF表达高度相关的基因
4. 湿实验验证:TF扰动实验 + RNA-seq
复杂的细胞状态(信号通路、环境刺激、非编码RNA、蛋白质复合物)控制着各种酶的活性(e.g. m6A Writer/Eraser/Reader),对RNA进行表观修饰、并通过这些修饰调控下游的剪接、翻译效率。
RNA的表观修饰(转录后修饰)有:m6A(剪接/翻译/降解),m5C(增强稳定性/影响核质转运),假尿苷(增强稳定性),A-to-I编辑。也可以通过 BS-seq / MeDIP-seq 获得甲基化修饰位点。RNA的表观修饰会影响各种调控蛋白/miRNA(e.g.与RNA互补)的结合效率,e.g. 加速招募mRNA降解相关蛋白/miRNA(mRNA稳定性),招募翻译起始因子/增强核糖体结合(mRNA翻译效率),于外显子-内含子交界处招募剪接因子,招募转运蛋白 ...
一般可以从 RNA-seq 中获取可变剪接事件 (rMATS)、其突变信息(ANNOVAR)。 关于其调控机制,可通过 SpliceAid 数据库预测剪接因子的结合位点,或使用 CLIP-seq (蛋白-RNA互作) 验证剪接因子(e.g. SRSF1)的结合位点(即调控剪接的顺式元件)。
翻译调控(RNA -> Peptide Translation)的研究。Ribo-seq 可获得与核糖体结合的RNA片段(正在翻译),其定位(ribosome footprints)的密集程度暗示翻译的效率:Translation Efficiency = Ribo-binding Reads (Ribo-seq) / mRNA reads (RNA-seq)
关于 Ribo-seq,可以联合其它组学:
- 翻译效率是否正常? --- (active mRNA) v.s. (蛋白组)
- mRNA稳定但翻译抑制?寻找相应的转录后修饰 --- (active mRNA) v.s. (RNA-seq / BS)
示例:一个研究问题
Alternative Promoter --> TSS 变化 --> pre-mRNA 变化,如何影响翻译效率?
- 长/短/序列不同的5'UTR --> 新增/减少 蛋白结合位点
- 可变剪接 --> 不同的外显子 --> 蛋白结构/功能改变
- 降解/翻译起始/.... 参考上文 ‘表观修饰’ 段
- 在上游增加 ORF,翻译后核糖体脱离,抑制主ORF的翻译
- 一些可行的ML工具
Single-Cell & Spatial
| -- | 说明 | -- |
|---|---|---|
| Single_Cell | Reads 带有 Cell Barcode 和 UMI | -- |
| Spatial | Reads 带有空间信息 | -- |