HiC
示意图
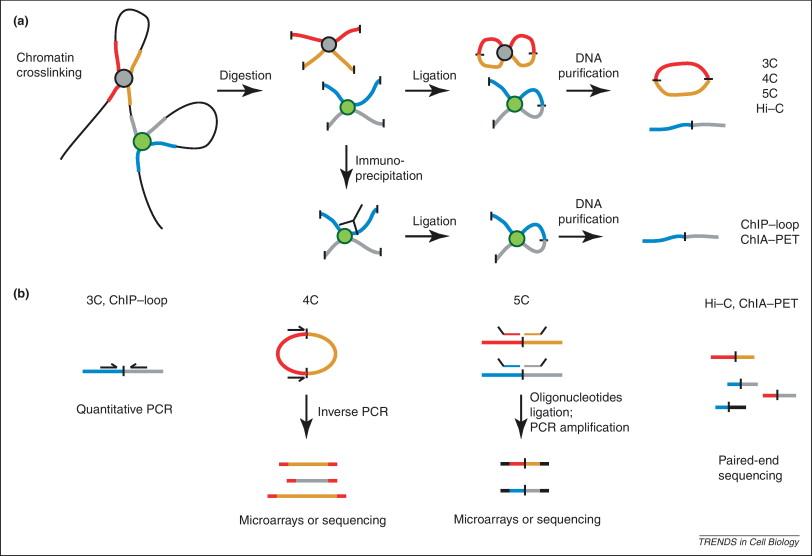
HiC相比3/4/5C可以做到全基因组范围的互作;将DNA用蛋白交联固定后,水解得到蛋白周边的DNA片段,连接DNA片段的末尾。如此,一个测序Interval中,前半段与后半段将会被map到基因组中两个不同的位置,提示这两个位置在三维空间中接近。
概念
真核生物的染色质空间组织有如下三个关键层级结构:(“增强子在A区室的TAD内通过Loop接触启动子”)
-
Compartments (区室) --- 全基因组范围、转录状态的活跃(A)和沉默(B)区室
- 1~10 Mb
- 成因:组蛋白修饰(e.g. A区室中H3K36me3富集)
- 意义:划分全局转录活跃与沉默区域
- 动态响应发育或环境信号(X染色体失活时,整条染色体从A区室转为B区室)
-
TADs (拓扑关联域) --- 局部互作频繁的连续区域、内部基因调控相对独立
- 0.1~1 Mb
- 成因:Cohesin介导的DNA环挤出(loop extrusion)被边界CTCF位点阻挡,形成稳定域结构
- 意义:限制调控元件的作用范围、隔离不同染色质状态(增强子通常只能激活同一TAD内的基因);TAD边界破坏可能导致Loops跨越原边界,引发异常基因表达
- 辅助验证:CRISPR删除边界序列,观察基因表达变化;ChIP-seq检测CTCF/Cohesin在边界的富集
- 边界相对保守,内部互作随细胞状态动态变化
-
Loops (染色质环) --- 远距离精准互作形成的环
- 几十kb ~ 几Mb
- 成因:Cohesin复合物将两个CTCF位点拉近形成环(e.g. 增强子-启动子互作)
- 意义:介导远端调控元件与目标基因的直接互作
- 高度动态/快速响应信号
CTCF位点是指CTCF(CCCTC-binding factor)蛋白结合的特定DNA序列,基因组中数量众多(约有15000个)
Gene Reguation Pipeline
最常用的工具集可能是Juicer与Hic-pro,建议直接问 Deepseek “AAA 如何使用”
| 工具 | 用途 | -- |
|---|---|---|
| HiC-Pro | fq生成交互矩阵 | -- |
| Juicer | fq生成交互矩阵,TAD/Loop | .hic格式与可视化工具 Juicebox 无缝集成 |
| Cooler | 格式转换,矩阵缩放/归一化 | 对于.cool格式,HiGlass支持可视化,cooltools支持TAD/diff |
| cworld-dekker | 主要下游分析 | 输入矩阵格式:bin1 bin2 count |
| HiCExplorer | bam生成交互矩阵,主要下游分析 | hicPlotxxx值得一试,--BED H3K27ac_peaks.bed 可叠加 Chip-seq peak |
主要下游分析:矩阵缩放/归一化,TAD/Loop,AB Compartment(PCA), 差异检测,热图
练习数据:表观遗传数据网站 ENCODE ,或各类 4DN DCIC workshop,推荐2018-bootcamp教程以及其提供的数据
常见格式
.hic: 二进制,Juicer系列的互作矩阵.cool/ 多分辨率.mcool: 基于 HDF5,包含 bins(基因组区间)、pixels(交互对)、weights(归一化系数).matrix (HiC-Pro)稀疏矩阵
bin1 bin2 count
1 1 50
1 2 20
.pairs可通过pairtools处理
## 注释行(基因组版本、格式说明)
#columns: readID chr1 pos1 chr2 pos2 strand1 strand2
read1 chr1 100000 chr2 200000 + -
-
.expected (Cooler): 归一化后的期望交互频率(用于比较) -
.loops / .domains(.bedpe): TAD/Loop区域chr1 1000000 1050000 chr1 2000000 2050000 0.05
4dn-dcic 提供的工具 hic2cool 可以进行 .hic → .cool 转换,而 cooler load/cload 可以将一些其它各种转换为 .cool
Preprocess -- 获得:互作矩阵
准备好 refGenome,随后将reads比对(bwa men --SP5M)。过滤、得到 Valid Pairs 后,将基因组拆为按不同分辨率的bins,得到互作矩阵(Contact Map)
HICUP(hicup_mapper), Juice (bwa), hiclib, HiC-Pro (bowtie2) 打包了这个流程,且都对bwa或bowtie2进行了优化设置。
限制性酶切位点来自实验设计,一些示例
背景:一般而言,对于一组PE reads,R1与R2将各自比上某个区域;但是,有时一些单侧Read过长、跨越了link site,如此它可能会匹配到两个区域。
除了常规的QC与Trimming,可以使用 HiCUP 修正一些跨越link site的单侧reads:
- 指定Type Ⅱ限制性内切酶切割参考基因组,生成整个参考基因组的酶切位点文件
- 通过酶切位点文件得到link site的序列,对跨越link site的单侧reads去除link site后的片段
- mapping,使用bowtie2
- 参照酶切位点文件,去除常见错误模式的HiC片段
- 去除PCR重复
A/B compartment
Idea: A/B 区室的位置有分隔,在PC1上会有明显区分
步骤:互作矩阵 --> z-scale --> PCA(取PC1),其(正/负)=>(A/B),正负phasing需要加上表观信息进行判断(Chip-seq数据)
验证:A compartment 区域应与高表达基因共定位,且富集 H3K27ac、DNase I 敏感位点
建议分辨率:50 kb - 1 Mb 哺乳动物,10-50 kb 果蝇
用途一:物种间比较 compartment 区域的保守性
- 方法一:Juicer 提取互作矩阵 + sklearn PCA (来自Deepseek,待验证)
# juicer_tools dump <observed/oe> <hicFile(s)> <chr1>[:x1:x2] <chr2>[:y1:y2] <BP/FRAG/分辨率 binsize> [outfile]
juicer_tools dump observed KR input.hic chr1 chr1 BP 100000 chr1_matrix.txt
python -c "
import numpy as np;
from sklearn.decomposition import PCA;
matrix = np.loadtxt('chr1_matrix.txt');
pca = PCA(n_components=1);
pc1 = pca.fit_transform(matrix).flatten();
np.savetxt('pc1.txt', pc1, fmt='%f');
"
paste hg38.chrom.sizes pc1.txt | awk '{print $1"\t"$2"\t"$3"\t"($4>0?"A":"B")}' > compartments.bed
- 方法二:cooltools 可自动 phasing
## pip install cooltools
## <input.cool> 若有分辨率则需指定,e.g. <input.mcool::resolutions/100000>
## ignore-diags 忽略前两个对角线(避免局部交互干扰)
## phasing-track genes.bed 利用基因密度文件自动校正方向(确保 A=活跃)
## Output: AB.eigenvector.bedGraph:PC1 值 (正/负)=>(A/B) -----
cooltools eigs-cis <input.cool> --phasing-track None --out-prefix AB --n-eigs 1 --ignore-diags 2
# awk '{print $1"\t"$2"\t"$3"\t"($4>0?"A":"B")}' AB.eigenvector.bedGraph > AB.bed
- 其它工具:homer, HiCExplorer, cworld-dekker
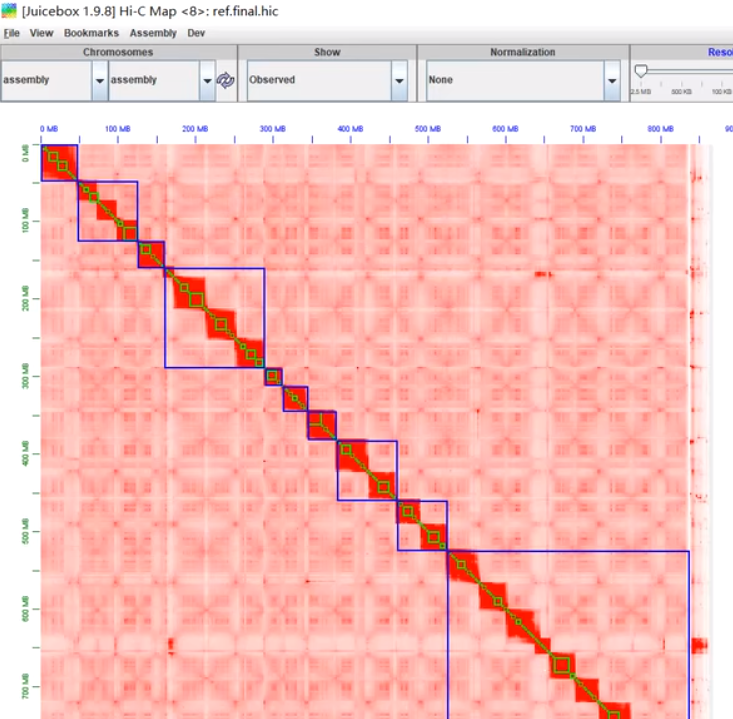
TAD
流程:互作矩阵 --> TAD边界鉴定(.bed) --> TAD图
建议分辨率:10-50 kb 哺乳动物,1-10 kb 果蝇
验证:边界通常富集CTCF/Cohesin(ChIP-seq验证),富集活跃调控元件(启动子/增强子)
用途一:跨物种/细胞类型比较边界位置(如phyloP评分)
用途二:边界破坏可能导致疾病(如癌症基因组重排)
若多种方法得到的边界不一致,可取其交集 bedtools intersect -a method1.bed -b method2.bed > consensus_boundaries.bed
-
方向性指数(Directionality Index, DI)
- DI = (binA 上游的交互总和 - 全局期望)/标准差 - (binA 上游的交互总和 - 全局期望)/标准差
- TAD边界处的交互会呈现不对称性(一侧高、一侧低),即:DI值的极值点(峰或谷)对应TAD边界
- 工具:hicFindTADs(HiCExplorer),cworld-dekker
-
- IS = log2(窗口内交互总和/全基因组背景交互)
- 通过滑动窗口计算局部交互的绝缘强度,边界处绝缘分数最低(交互被阻断),即:局部最小值对应TAD边界
- 工具:cooltools insulation
-
几何特征:矩阵三角形检测
- 归一化矩阵(KR/VC) --> 滑动窗口检测交互强度的锐减拐点 --> 过滤假阳性(如技术噪声)
- 工具:juicer_tools arrowhead
-
其它:Graph 聚类分割,HMM,DL 多模态数据整合预测TAD(e.g. CTCF、组蛋白修饰)
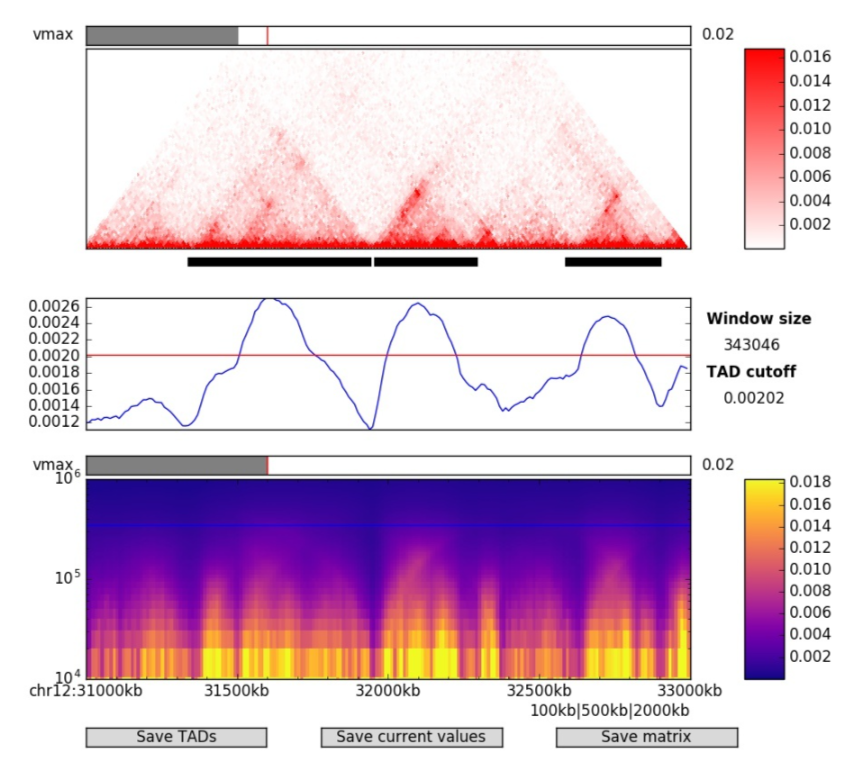
Loop
流程:互作矩阵 --> (显著高的交互频率,基于不同统计模型) --> Loop 锚点对(.bedpe 文件,包含 Loop 锚点坐标和交互强度)
原理:(每个候选位点的) Local Interaction Enrichment = 实际观测的交互值 / 基于距离衰减模型的期望交互值(通常通过基因组距离拟合)
建议分辨率:5kb - 10kb 哺乳动物
验证:边界通常富集CTCF/Cohesin(ChIP-seq验证),Loop 锚点(anchor)两侧交互强度对称,距离范围 20 kb - 2 Mb(哺乳动物)
-
juicer_tools hiccups- 在不同分辨率下(如 5kb/10kb
-r 5000,10000)检测局部交互峰 - 一定记得归一化
-k KR
- 在不同分辨率下(如 5kb/10kb
-
Fit-Hi-C
- 基于 负二项分布 建模交互频率,检测显著富集的交互对
- 低分辨率数据(>10 kb)或稀疏矩阵
Scaffolding and Phasing
HiC可用来辅助进行染色体级别基因组的组装;不过HiC组装的染色图其实存在大量错误,需要用更精细的遗传连锁图谱进行纠正
| 工具 | 流程 | 说明 |
|---|---|---|
| LACHESIS | 1.根据contact将reads分组;2.组内reads ordering、组装;3.contigs orient | 经典,但停止更新 |
| 3D-DNA + Juicer | 1.切割与HiC数据相悖的contigs;2.Juicer 得到Hi-C maps;3.3D-DNA 根据map重新链接;4.juicerbox 手动矫正 | 二倍体效果最佳 |
| All-HiC | -- | 针对多倍体和高杂合度的情况 |
| chromap+YaHS | 1.chromap快速mapping;2.YaHS scaffolding | 更快,似乎排序更准确 |
- 其余:HiFiasm 可直接基于HiC数据进行组装,上表只是辅助
- 评估方法:准确度(how?),挂载率(草图中有多少比例的base被包含在染色体中)
Meta HiC
hicSPAdes, HiCBin等使用HiC技术辅助Binning
参考
A/B 染色质区室: https://cloud.tencent.com/developer/article/1556901
PMC:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4490074/
HiC辅助组装:https://maimengkong.com/m/?post=1178