BLAST
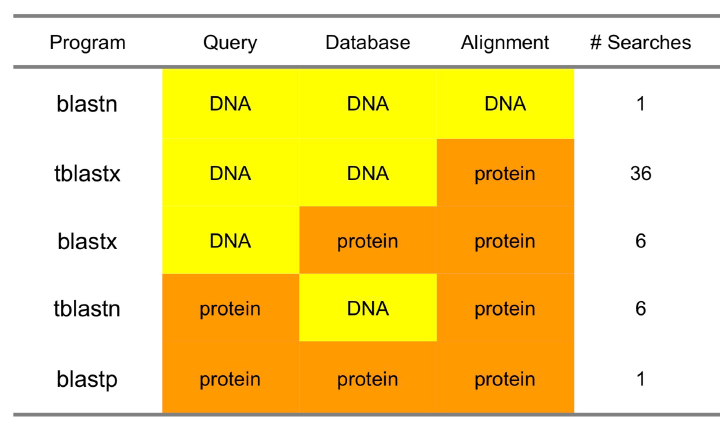
注:由于不确定翻译起始,nt-->aa 有6种可能
faa: protein
fna: nucleotide
另:各种参数的选择建议参考同类论文
Install
conda install -c bioconda blast
conda install -c bioconda diamond
apt,conda,release 都可以安装;只是我的wsl中diamond只能成功安装v0.9
BLAST+ package
- search tools: blastn, blastp, blastx, tblastx, tblastn, psiblast, rpsblast, rpstblastn
- database tools: makeblastdb, blastdb_aliastool, makeprofiledb, blastdbcmd
makeblastdb
makeblastdb -in DB.fna -input_type fasta -dbtype nucl -out DB_nt -parse_seqids
makeblastdb -in DB.faa -input_type fasta -dbtype prot -out DB_aa -parse_seqids
## -dbtype nucl/prot/guess
## without -parse_seqids:
### DB_nt.nhr DB_nt.nin DB_nt.nsq
## with -parse_seqids:
### DB_nt.nhr DB_nt.nin DB_nt.nsq DB_nt.nog DB_nt.nsd DB_nt.nsi
blastdbcmd
## date & info
blastdbcmd -db DB_nt -info | less
## back to FASTA
blastdbcmd -db DB_nt -entry all | less
blastn
blastn -query in.fna -db DB_nt -out DB_nt.m8 -outfmt 6 -evalue 1e-5 -max_target_seqs 5 -num_threads 20
-outfmt可以控制输出列,详情见下文
-outfmt '6 qseqid sseqid qstart qend sstart send evalue'
对于短序列(e.g. 20bp),建议
-task blastn-short -word_size 4 -evalue 1
## -task: 'blastn' 'blastn-short' 'dc-megablast'
psiblast
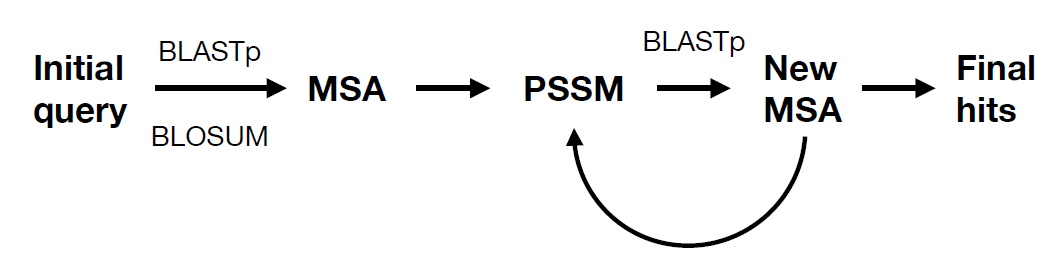
- 迭代PSSM矩阵、不断发现新的match
- 一次为一条蛋白序列计算PSSM
- 对于SWISS数据库需要query序列>15bp,否则需要试试更大的数据库(e.g. NR)
- 生成 L*20维 矩阵,但长度不固定(L为input长度);后续作为机器学习输入前需要进行信息提取
psiblast -query single_in.faa -db DB_aa -out pssm.m8 -outfmt 6 -evalue 1e-3 -num_iterations 3 -out_ascii_pssm pssm.txt
Diamond
Diamond的blastp似乎比BLAST+更快,于是和蛋白数据库相关的一般用它;注意:无root权限时运行比对命令需要指定临时文件夹(需创建)
--tmpdir /tmp/mytmpdir/
makedb
Diamond只支持blastp、blastx;它俩使用protein DB
diamond makedb --in DB.faa --db DB_aa.dmnd
## db info
diamond dbinfo -d DB_aa.dmnd |less
## back to FASTA
diamond getseq -d DB_aa.dmnd |less
blastp
mkdir temp_dir
diamond blastp -d DB_aa.dmnd -q in.faa -o out_p.m8 -f 6 -e 1e-5 -k 5 -b 2 -t ./temp_dir
-f 6 (outformat 6: BLAST table format)
-k 10 (max-target-seqs,设置每个query比对结果的最大匹配数目)
-b sequence block size in billions of letters (default=2.0)
blastx
diamond blastx -d DB_aa.dmnd -q in.fna -o out_x.m8 -f 6 -e 1e-5 -k 5 -b 2 -t ./temp_dir \
--threads 20 --quiet --id 10 --subject-cover 50 --query-cover 50
Output Format
PSSM
Last position-specific scoring matrix computed, weighted observed percentages rounded down, information per position, and relative weight of gapless real matches to pseudocounts
A R N D C Q E G H I L K M F P S T W Y V A R N D C Q E G H I L K M F P S T W Y V
1 M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 0 -2 -1 -1 -1 -1 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0.00 0.00
2 M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 0 -2 -1 -1 -1 -1 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0.00 0.00
3 F -2 -3 -3 -3 -2 -3 -3 -3 -1 0 0 -3 0 6 -4 -2 -2 1 3 -1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0.00 0.00
4 G 0 -2 0 -1 -2 -2 -2 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0.00 0.00
5 G 0 -2 0 -1 -2 -2 -2 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0.00 0.00
6 K -1 2 0 -1 -3 1 1 -2 -1 -3 -2 4 -1 -3 -1 0 -1 -3 -2 -2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0.00 0.00
7 S 1 -1 1 0 -1 0 0 0 -1 -2 -2 0 -1 -2 -1 4 1 -3 -2 -2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0.00 0.00
8 M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 0 -2 -1 -1 -1 -1 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0.00 0.00
9 G 0 -2 0 -1 -2 -2 -2 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0.00 0.00
......
m8
常用的-outfmt 6输出为:
X17276.1 X17276 100.000 556 0 0 1 556 1 556 0.0 1027
X51700.1 X53699 100.000 437 0 0 1 437 1 437 0.0 808
X51700.1 X51700 100.000 437 0 0 1 437 1 437 0.0 808
X68321.1 X68321 100.000 1512 0 0 1 1512 1 1512 0.0 2793
.....
1 2 3 4 5 6 7 8 9 10 11 12
qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore
Details
blastn -help
-outfmt <String>
alignment view options:
0 = Pairwise,
1 = Query-anchored showing identities,
2 = Query-anchored no identities,
3 = Flat query-anchored showing identities,
4 = Flat query-anchored no identities,
5 = BLAST XML,
6 = Tabular,
7 = Tabular with comment lines,
8 = Seqalign (Text ASN.1),
9 = Seqalign (Binary ASN.1),
10 = Comma-separated values,
11 = BLAST archive (ASN.1),
12 = Seqalign (JSON),
13 = Multiple-file BLAST JSON,
14 = Multiple-file BLAST XML2,
15 = Single-file BLAST JSON,
16 = Single-file BLAST XML2,
17 = Sequence Alignment/Map (SAM),
18 = Organism Report
Options 6, 7, 10 and 17 can be additionally configured to produce
a custom format specified by space delimited format specifiers.
The supported format specifiers for options 6, 7 and 10 are:
qseqid means Query Seq-id
qgi means Query GI
qacc means Query accesion
qaccver means Query accesion.version
qlen means Query sequence length
sseqid means Subject Seq-id
sallseqid means All subject Seq-id(s), separated by a ';'
sgi means Subject GI
sallgi means All subject GIs
sacc means Subject accession
saccver means Subject accession.version
sallacc means All subject accessions
slen means Subject sequence length
qstart means Start of alignment in query
qend means End of alignment in query
sstart means Start of alignment in subject
send means End of alignment in subject
qseq means Aligned part of query sequence
sseq means Aligned part of subject sequence
evalue means Expect value
bitscore means Bit score
score means Raw score
length means Alignment length
pident means Percentage of identical matches
nident means Number of identical matches
mismatch means Number of mismatches
positive means Number of positive-scoring matches
gapopen means Number of gap openings
gaps means Total number of gaps
ppos means Percentage of positive-scoring matches
frames means Query and subject frames separated by a '/'
qframe means Query frame
sframe means Subject frame
btop means Blast traceback operations (BTOP)
staxid means Subject Taxonomy ID
ssciname means Subject Scientific Name
scomname means Subject Common Name
sblastname means Subject Blast Name
sskingdom means Subject Super Kingdom
staxids means unique Subject Taxonomy ID(s), separated by a ';'
(in numerical order)
sscinames means unique Subject Scientific Name(s), separated by a ';'
scomnames means unique Subject Common Name(s), separated by a ';'
sblastnames means unique Subject Blast Name(s), separated by a ';'
(in alphabetical order)
sskingdoms means unique Subject Super Kingdom(s), separated by a ';'
(in alphabetical order)
stitle means Subject Title
salltitles means All Subject Title(s), separated by a '<>'
sstrand means Subject Strand
qcovs means Query Coverage Per Subject
qcovhsp means Query Coverage Per HSP
qcovus means Query Coverage Per Unique Subject (blastn only)
When not provided, the default value is:
'qacc sacc pident length mismatch gapopen qstart qend sstart send evalue
bitscore', which is equivalent to the keyword 'std'
The supported format specifier for option 17 is:
SQ means Include Sequence Data
Default = `0'
diamond help
--outfmt (-f) output format
0 = BLAST pairwise
5 = BLAST XML
6 = BLAST tabular
100 = DIAMOND alignment archive (DAA)
101 = SAM
Value 6 may be followed by a space-separated list of these keywords:
qseqid means Query Seq - id
qlen means Query sequence length
sseqid means Subject Seq - id
sallseqid means All subject Seq - id(s), separated by a ';'
slen means Subject sequence length
qstart means Start of alignment in query
qend means End of alignment in query
sstart means Start of alignment in subject
send means End of alignment in subject
qseq means Aligned part of query sequence
sseq means Aligned part of subject sequence
evalue means Expect value
bitscore means Bit score
score means Raw score
length means Alignment length
pident means Percentage of identical matches
nident means Number of identical matches
mismatch means Number of mismatches
positive means Number of positive - scoring matches
gapopen means Number of gap openings
gaps means Total number of gaps
ppos means Percentage of positive - scoring matches
qframe means Query frame
btop means Blast traceback operations(BTOP)
staxids means unique Subject Taxonomy ID(s), separated by a ';' (in numerical order)
stitle means Subject Title
salltitles means All Subject Title(s), separated by a '<>'
qcovhsp means Query Coverage Per HSP
qtitle means Query title
Default: qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore
参考
BLAST: https://blast.ncbi.nlm.nih.gov/Blast.cgi
Diamond: https://github.com/bbuchfink/diamond
BLAST比对结果: https://www.jianshu.com/p/9aa1a131473e
比对软件默认eval: https://www.jianshu.com/p/ea8218a3ff18
PSSM: https://blog.csdn.net/cbb_ft/article/details/124623766